




La enfermedad por hígado graso no alcohólico es un espectro patológico que va desde la simple esteatosis hasta la esteatohepatitis, en ausencia de consumo de alcohol en cantidades perjudiciales, y se considera la manifestación hepática del síndrome metabólico. Estudios recientes indican que se relaciona estrechamente con la enfermedad cardiovascular, sobre todo con el engrosamiento de la capa íntima-media de la arteria carótida, como manifestación morfoestructural de la presencia de ateromatosis subclínica. Por tanto, el manejo correcto de la enfermedad por depósito graso no alcohólico permitirá modificar la historia natural de la enfermedad tanto hepática como aterosclerótica.
Non-alcoholic fatty liver disease encompasses a spectrum ranging from simple steatosis to steatohepatitis without excess alcohol intake and is considered to be the hepatic manifestation of metabolic syndrome. Recent studies indicate that non-alcoholic fatty liver disease is closely related to cardiovascular disease, especially to thickening of the intima-media layer of the carotid artery, as the morphostructural manifestation of the presence of subclinical atheromatosis. Therefore, the correct management of non-alcoholic fatty liver disease would allow the natural history of both the liver disease and the atherosclerosis to be modified.
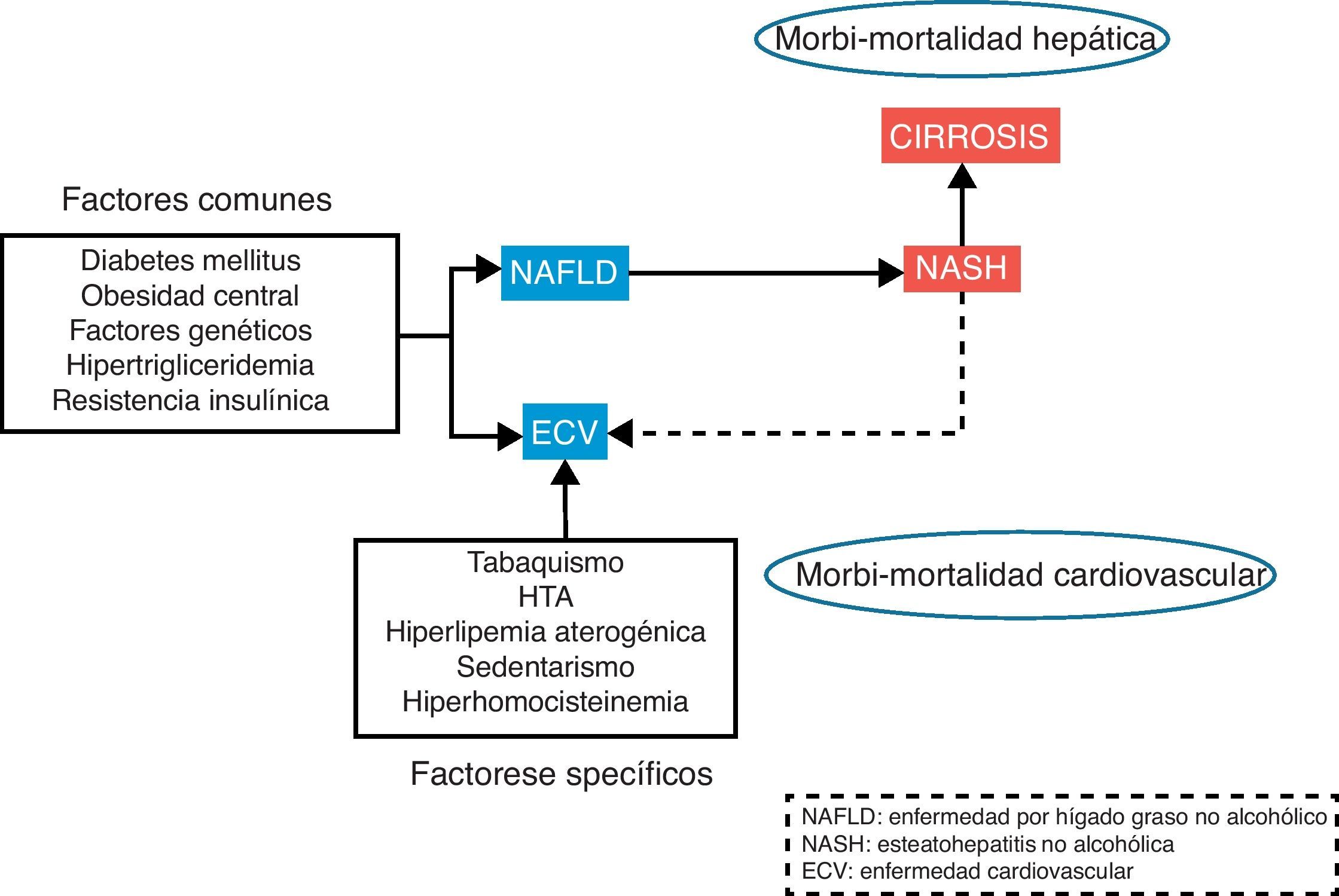
Las enfermedades cardiovasculares continúan siendo la primera causa de muerte e incapacidad en el mundo. La manifestación inicial en el desarrollo de la enfermedad cardiovascular es la aterosclerosis. La aparición de la placa de ateroma es un proceso complejo que se desarrolla como consecuencia de la interacción entre la carga genética, los estímulos externos y las diferentes alteraciones metabólicas. La enfermedad hepática por depósito de grasa no alcohólica (NAFLD) se presenta como un problema de salud cada vez más importante en la población general, tanto por su alta incidencia como por su morbimortalidad. La elevada incidencia y prevalencia se debe a una estrecha relación con el síndrome metabólico1, de hecho existen una serie de factores de riesgo comunes como la diabetes, obesidad, resistencia a la insulina o hiperlipidemia implicadas en el desarrollo tanto de la enfermedad cardiovascular como NAFLD (fig. 1). Además, esta enfermedad hepática asociada a trastornos metabólicos se asocia a morbimortalidad, tanto cardiovascular2 como hepática3, incluido el desarrollo de cirrosis e, incluso, hepatocarcinoma.
Aterosclerosis
La aterosclerosis es un síndrome caracterizado por el depósito e infiltración de sustancias lipídicas en las paredes de las arterias de mediano y grueso calibre. Es un proceso inflamatorio crónico en la pared arterial, cuyo mecanismo fisiopatológico estriba en el aumento de susceptibilidad de las lipoproteínas de baja densidad (LDL) a la oxidación por radicales libre. A estas moléculas oxidadas se unen los monocitos y este conjunto se adhiere a la capa íntima arterial, provocando una respuesta inflamatoria que induce la transformación de los monocitos en macrófagos. En situaciones de exceso de LDL, los macrófagos son incapaces de eliminarlas, convirtiéndose en células espumosas cargadas de colesterol que conforman la placa de ateroma4. Este proceso es lento, desarrollándose de manera silente y estando muy avanzado cuando aparecen los síntomas. Por ello, el diagnóstico precoz ha de hacerse en la fase de ateromatosis subclínica, cuando es posible modificar la evolución de la enfermedad y disminuir el riesgo cardiovascular.
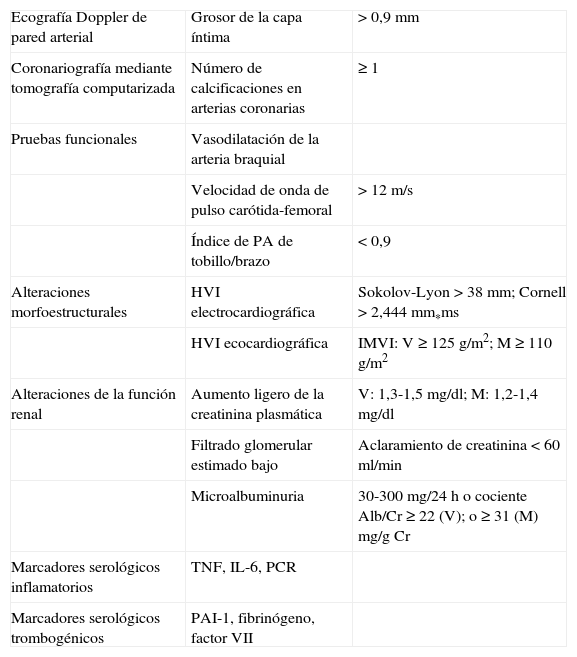
Se han empleado diversos métodos para la detección de la ateromatosis subclínica: a) la ecografía de la pared arterial ha sido la más empleada por su inocuidad, bajo coste y disponibilidad; mide el grosor de la capa íntima-media a nivel carotídeo, que es un indicador de enfermedad aterosclerótica en las arterias coronarias; los resultados obtenidos en la valoración de la aterosclerosis subclínica se han de corregir en función de la raza, el sexo y la edad5; b) la tomografía computarizada (TC) posibilita la detección de calcificaciones en las arterias coronarias; el número de calcificaciones es un marcador directo de la extensión de la aterosclerosis coronaria6; c) pruebas funcionales, como la valoración de la vasodilatación mediada por flujo en la arteria braquial permiten evaluar la disfunción endotelial, estrechamente relacionada con la incidencia y prevalencia de enfermedades cardiovasculares7. La velocidad de la onda de pulso carótida-femoral y el índice de presión arterial tobillo/brazo son también marcadores precoces de aterosclerosis subclínica; d) indicadores de análisis morfoestructural como la valoración de la hipertrofia ventricular izquierda, tanto electro como ecocardiográfica; e) alteraciones de la función renal, valorada por el aumento de la creatinina plasmática, descenso del filtrado glomerular y la microalbuminuria que pueden indicar la presencia de lesión orgánica subclínica8 (tabla 1); f) marcadores serológicos inflamatorios como la proteína C reactiva (PCR), la interleucina 6 (IL-6) y el factor de necrosis tumoral alfa (TNFα). La PCR se considera un predictor independiente de enfermedad coronaria, ya que acelera la aterosclerosis mediante el incremento de expresión del inhibidor de la activación del plasminógeno 1 (PAI-1), la adhesión de moléculas a las células endoteliales, el descenso de producción de óxido nítrico y el incremento de la captación de LDL9. La IL-6 ejerce su acción proaterosclerótica mediante la estimulación de la actividad PCR y la secreción de molécula de adhesión intercelular 1 (ICAM-1). De hecho, se ha demostrado asociación entre los valores de IL-6 y el engrosamiento de la capa íntima-media de la arteria carótida y, por tanto, con la aterosclerosis subclínica10. El TNFα aumenta la agregación plaquetaria y reduce la síntesis del óxido nítrico al inhibir la actividad de la oxido nítrico sintetasa endotelial11; g) marcadores serológicos trombogénicos como el aumento del fibrinógeno, el factor VII o PAI-1. El fibrinógeno ejerce su acción aumentando la viscosidad sanguínea, lo cual acelera la formación del trombo. El factor VII de la coagulación es esencial para la formación de trombos y se ha detectado mayor actividad en estados hipertrigliceridémicos. El PAI-1 es el principal inhibidor de la fibrinólisis, por lo que concentraciones elevadas de éste aumentan el riesgo protrombótico12.
Pruebas de lesión de órgano diana subclínicas
| Ecografía Doppler de pared arterial | Grosor de la capa íntima | > 0,9 mm |
| Coronariografía mediante tomografía computarizada | Número de calcificaciones en arterias coronarias | ≥ 1 |
| Pruebas funcionales | Vasodilatación de la arteria braquial | |
| Velocidad de onda de pulso carótida-femoral | > 12 m/s | |
| Índice de PA de tobillo/brazo | < 0,9 | |
| Alteraciones morfoestructurales | HVI electrocardiográfica | Sokolov-Lyon > 38 mm; Cornell > 2,444 mm*ms |
| HVI ecocardiográfica | IMVI: V ≥ 125 g/m2; M ≥ 110 g/m2 | |
| Alteraciones de la función renal | Aumento ligero de la creatinina plasmática | V: 1,3-1,5 mg/dl; M: 1,2-1,4 mg/dl |
| Filtrado glomerular estimado bajo | Aclaramiento de creatinina < 60 ml/min | |
| Microalbuminuria | 30-300 mg/24 h o cociente Alb/Cr ≥ 22 (V); o ≥ 31 (M) mg/g Cr | |
| Marcadores serológicos inflamatorios | TNF, IL-6, PCR | |
| Marcadores serológicos trombogénicos | PAI-1, fibrinógeno, factor VII |
HVI: hipertrofia ventricular izquierda; IMVI: índice de masa ventricular izquierda; M: mujeres; V: varones.
Los factores de riesgo clásicos, como la dislipidemia, la hipertensión arterial (HTA) y la diabetes mellitus tipo 2 (DM2) promueven la disfunción endotelial y una respuesta inflamatoria responsable del inicio, progresión y desarrollo de complicaciones de las lesiones vasculares. El aumento de colesterol total debido al incremento del cLDL es el principal factor de riesgo. La modificación oxidativa de las moléculas LDL produce la activación del endotelio y la retención de la propia molécula en la capa íntima de la arteria, iniciando una respuesta inflamatoria crónica en la pared arterial13. El aumento de moléculas LDL pequeñas y densas que depositan colesterol en las arterias, así como el incremento de triglicéridos y el descenso de colesterol unido a lipoproteínas de alta densidad (cHDL) es conocido como dislipidemia aterogénica14. La HTA es otro de los principales factores de riesgo cardiovascular por su habilidad para promover la hipertrofia ventricular. El endotelio está expuesto al estrés mecánico creado por el flujo sanguíneo al circular a alta presión, es decir, por fuerzas de cizallamiento que rompen el endotelio de las arterias, pudiendo depositarse calcio y ácidos grasos en las zonas debilitadas15. Los sujetos con DM2 presentan una evolución más acelerada de la enfermedad aterosclerótica16 debido a la existencia de un estado de hiperinsulinemia por la resistencia a la insulina que, por un lado, aumentaría la síntesis de ácidos grasos libres y, por otro, incrementaría los procesos oxidativos e inflamatorios, favoreciendo la formación de placas de ateroma a través de la oxidación de LDL17,18. El tabaquismo, el sedentarismo y la ausencia de dietas cardiosaludables (dietas ricas en grasas saturadas), así como ciertas alteraciones genéticas, completan el listado de factores de riesgo de aterosclerosis subclínica y enfermedad cardiovascular.
Síndrome metabólicoEl síndrome metabólico es un estado patológico donde tiene lugar la conjunción de diferentes alteraciones que aumentan la posibilidad de presentar una enfermedad cardiovascular. Su aparición puede ser simultánea o secuencial. La prevalencia de síndrome metabólico varía entre zonas geográficas, grupos étnicos y edad. Se estima una prevalencia del 20% en la población occidental y del 75% en sujetos con DM219. Los parámetros incluidos en la definición del síndrome metabólico son la obesidad central, la dislipidemia, la HTA y la alteración del metabolismo glucídico. Se acepta que la resistencia a la insulina es el mecanismo fisiopatológico subyacente; un estado de hiperinsulinemia necesario para controlar la glucemia pero también responsable del aumento de la producción hepática de lipoproteínas de muy baja densidad (VLDL) y triglicéridos y de la puesta en marcha del proceso de la aterosclerosis.
Enfermedad por hígado graso no alcohólicoLa NAFLD se considera la manifestación hepática del síndrome metabólico20. Su incidencia está aumentando en los últimos años y puede encontrarse hasta en un 30% de la población general y hasta en un 70% en individuos obesos o con DM221. La etapa más temprana es la esteatosis hepática simple, que consiste en la acumulación de triglicéridos en el citoplasma de los hepatocitos, en forma de vacuolas lipídicas. La esteatosis hepática suele ser autolimitada, pero en ocasiones (5% aproximadamente) puede progresar a esteatohepatitis no alcohólica (NASH). La NASH presenta, como característica definitoria, infiltrado inflamatorio, además de lesión en el hepatocito (balonización y muerte celular) y depósito de colágeno (fibrosis). Tras la instauración de NASH, hasta un 30% de pacientes pueden desarrollar cirrosis en 10 años y, en última instancia, hepatocarcinoma22.
La esteatosis hepática es consecuencia de un desequilibrio entre la acumulación y la eliminación de triglicéridos, los cuales se mantienen unidos a ácidos grasos libres (FFA). La procedencia de estos FFA es la dieta, la síntesis de novo y el tejido adiposo. La dieta rica en hidratos de carbono provoca un aumento en las concentraciones de insulina que conlleva la síntesis de novo de FFA (gracias a acetilcoenzima A) al sobreexpresar SREBP-1c (vía AKT2, LXR y mTOR) y ChREBP. Por el contrario, durante el ayuno los niveles de insulina descienden y el glucagón se encarga de estimular la hidrólisis de triglicéridos en los adipocitos (mediante la lipasa adiposa de triglicéridos [ATGL]), con el posterior transporte de FFA al hígado. En el hígado, los FFA pueden ser oxidados en la mitocondria, incorporados a los triglicéridos o almacenados y secretados en forma de VLDL23. Sin embargo, la activación de SREBP-1c inhibe la oxidación de FFA y contribuye a la menor formación de VLDL, por lo que se dificulta el transporte del exceso de triglicéridos hacia el torrente sanguíneo24. Cuando la síntesis de triglicéridos sobrepasa la producción de VLDL y su exportación, los triglicéridos se acumulan originando esteatosis hepática. La relación entre la obesidad y la esteatosis se ha puesto de manifiesto en numerosos estudios, y se explica, al menos en parte, por la producción de FFA en el tejido adiposo y la dieta rica en hidratos de carbono (en especial fructosa). La resistencia a la insulina, por otro lado, disminuye el efecto inhibitorio de la propia insulina sobre la producción de glucosa, mientras que el efecto estimulador sobre la lipogénesis es mantenido25. Además, el estrés oxidativo está incrementado y se produce inflamación en los hepatocitos, por la excesiva producción de especies reactivas de oxígeno y citocinas proinflamatorias (TNFα, IL-6, PCR), siendo estas esenciales para el desarrollo de NASH26. Por tanto, la obesidad y la resistencia a la insulina son los 2 factores más prevalentes de NAFLD.
La prueba de referencia de diagnóstico de NAFLD es la biopsia hepática, la cual muestra la acumulación de grasa en los hepatocitos, así como los diversos grados de inflamación y fibrosis (es la única prueba que puede discernir entre NAFLD y NASH). Los candidatos ideales para realizar este procedimiento serían aquellos que presentasen 2 de las 3 características del índice HAIR: hiperecogenicidad del parénquima hepático, resistencia a la insulina y/o elevación de transaminasas27. A menudo el diagnóstico de NAFLD suele hacerse mediante ecografía abdominal, donde podemos observar un aumento de ecogenicidad hepática, en ausencia de otras enfermedades (hepatitis C, enfermedad de Wilson, enfermedades autoinmunes, etc.) o ingesta de alcohol por el aumento de transaminasas hepáticas (la alanino aminotransferasa [ALT] se correlaciona mejor con la presencia de esteatosis que la aspartato aminotransferasa [AST] o la gammaglutamil transpeptidasa [GGT]). No obstante, estas alteraciones aisladas tienen una baja seguridad diagnóstica, lo que disminuye el impacto de los estudios clínicos y epidemiológicos.
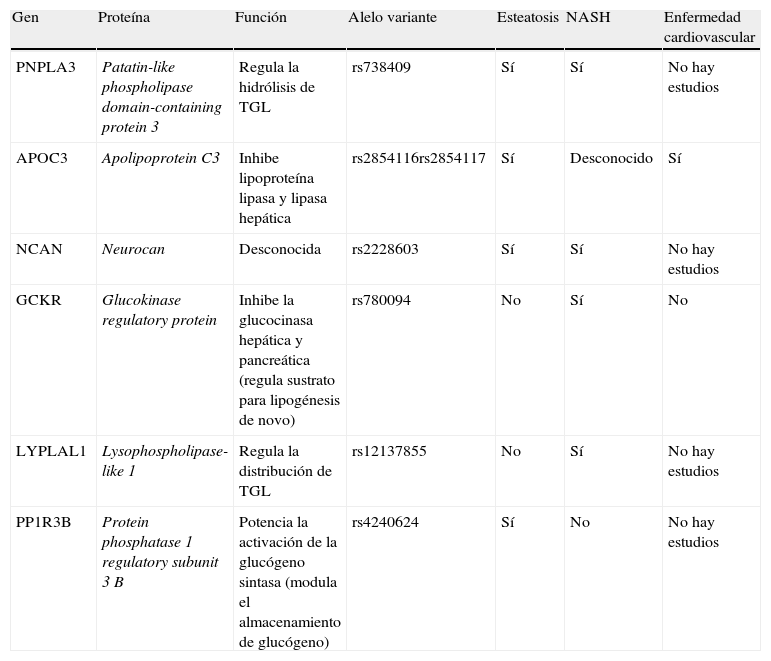
Función de la genética en la enfermedad por hígado graso no alcohólicoIndividuos con características similares en cuanto a obesidad y resistencia insulínica presentan, sin embargo, variaciones en el contenido de grasa hepática, lo que fundamenta la presencia de factores genéticos (tabla 2). Entre ellos, destacan APOC3 y PNPLA3.
Factores genéticos predisponentes de enfermedad por hígado graso no alcohólico y su relación con la enfermedad cardiovascular
| Gen | Proteína | Función | Alelo variante | Esteatosis | NASH | Enfermedad cardiovascular |
| PNPLA3 | Patatin-like phospholipase domain-containing protein 3 | Regula la hidrólisis de TGL | rs738409 | Sí | Sí | No hay estudios |
| APOC3 | Apolipoprotein C3 | Inhibe lipoproteína lipasa y lipasa hepática | rs2854116rs2854117 | Sí | Desconocido | Sí |
| NCAN | Neurocan | Desconocida | rs2228603 | Sí | Sí | No hay estudios |
| GCKR | Glucokinase regulatory protein | Inhibe la glucocinasa hepática y pancreática (regula sustrato para lipogénesis de novo) | rs780094 | No | Sí | No |
| LYPLAL1 | Lysophospholipase-like 1 | Regula la distribución de TGL | rs12137855 | No | Sí | No hay estudios |
| PP1R3B | Protein phosphatase 1 regulatory subunit 3 B | Potencia la activación de la glucógeno sintasa (modula el almacenamiento de glucógeno) | rs4240624 | Sí | No | No hay estudios |
La apolipoproteína C3 (APOC3) es un componente proteico de las VLDL, cuya función es inhibir la lipoproteína lipasa y la lipasa hepática; como consecuencia, el consumo hepático de partículas ricas en triglicéridos es disminuido. Por tanto, el aumento de los niveles de APOC3 se traduce en el desarrollo de hipertrigliceridemia28. Esta molécula ha cobrado mucha importancia desde que Petersen et al. identificaron 2 polimorfismos de APOC3 (C-482T y T-455C) que la vinculaban a RI y NAFLD, con un incremento del 60% en los valores de triglicéridos en ayuno. Los autores concluyen que la presencia de estos polimorfismos aumentan sus valores plasmáticos, inhibiendo el aclaramiento de triglicéridos y conduciendo a un aumento en las concentraciones de quilomicrones, que son captados en el hígado y dan lugar al desarrollo de esteatosis29. Sin embargo, artículos posteriores se han mostrado contradictorios al respecto30,31. Por otra parte, diversos estudios indican que el aumento de la concentración sérica de APOC3 podría ser un factor predictivo de mortalidad cardiovascular32–34, incluso en la población diabética35.
PNPLA3La patatin-like phospholipase domain-containing 3 (PNPLA3) (también conocida como adiponutrina) es un miembro de la familia PNLPA. Son lipasas que participan en la hidrólisis de triglicéridos (por ejemplo, ATGL es PNPLA2). Es expresada en el tejido adiposo y en los hepatocitos (casi el 90% se sitúa en las vacuolas lipídicas), siendo regulada por la insulina a través de LXR y SREBP-1c36. Romeo et al.37 publicaron una variación en PNPLA3 (PNPLA3-148M) que estaba asociada a un incremento de la esteatosis hepática y de la inflamación hepática, pero no a resistencia insulínica. Los pacientes homocigotos para el alelo de riesgo tienen un riesgo dos veces mayor de acumulación de triglicéridos y podrían explicar la diferencia de frecuencia de esteatosis hepática que existe entre distintas razas38. Al igual que con APOC3, es una molécula que está siendo ampliamente investigada y se ha relacionado con el desarrollo de esteatosis y fibrosis en la hepatitis crónica C39, con mayor riesgo de daño hepático en la enfermedad hepática alcohólica40 y con mayor fibrosis hepática en individuos con DM241. Hasta la fecha no existen estudios que investiguen la relación entre la PNPLA3 y la enfermedad cardiovascular.
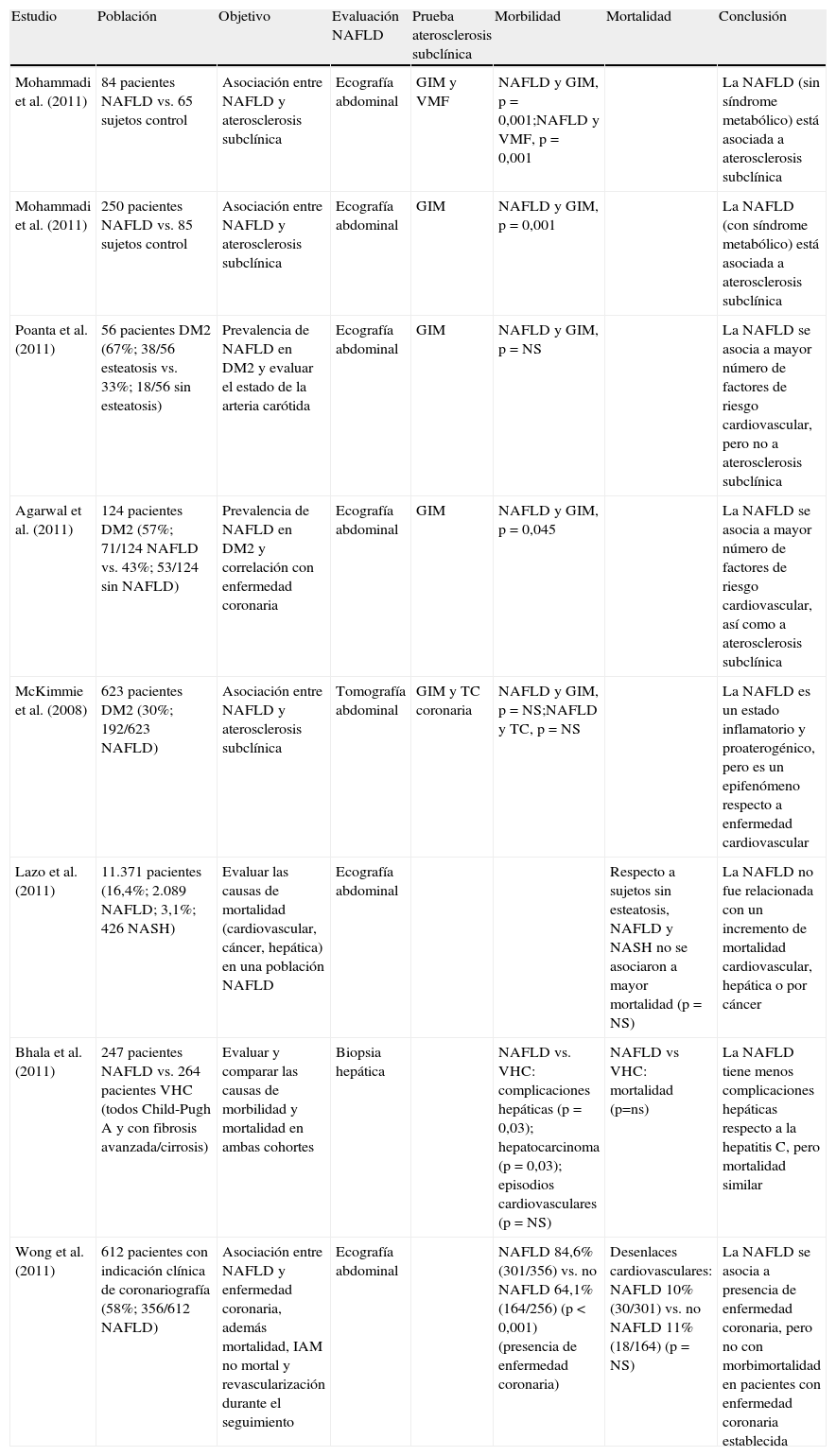
Influencia de la enfermedad por hígado graso no alcohólico en la enfermedad cardiovascularEnfermedad por hígado graso no alcohólico y aterosclerosis subclínicaEstudios recientes demuestran una estrecha relación entre la presencia de NAFLD y la aterosclerosis subclínica (tabla 3). Mohammadi et al.42 examinaron el engrosamiento de la capa íntima-media de la arteria carótida y la vasodilatación mediada por flujo en la arteria braquial en 84 pacientes con diagnóstico de NAFLD (mediante ecografía) y 65 sujetos control y concluyeron que las diferencias en los cambios morfológicos y funcionales eran significativas (p = 0,001). Los sujetos incluidos en el estudio eran menores de 45 años y no presentaban características propias de síndrome metabólico, por lo que los autores concluyen que la influencia de NAFLD es una variable independiente asociada a la aterosclerosis subclínica en la población de mediana edad sin síndrome metabólico. En otro artículo publicado por el mismo autor se compararon 250 pacientes con diagnóstico de NAFLD (mediante ecografía) con 85 sujetos control aleatorizados según edad y sexo, en relación con el grosor de la capa íntima-media carotídea y la presencia de placas ateroscleróticas en dicha arteria. En este caso sí se trata de individuos con síndrome metabólico y tras el análisis multivariante, la presencia de NAFLD se relacionó con el engrosamiento de la capa íntima-media (p = 0,001), independientemente de otros factores de riesgo aterogénicos asociados con el síndrome metabólico43. Se han publicado estudios que miden el impacto de NAFLD en la historia natural de la DM2. Poanta et al. publicaron un estudio de 56 sujetos con DM2 no complicada, a quienes mediante ecografía se clasificaba como esteatosis hepática (38/56; 67%) frente a no esteatosis (18/56; 33%). Aquellos que presentaban esteatosis hepática mostraban un índice mayor de factores de riesgo cardiovascular (índice de masa corporal, HTA, concentración de triglicéridos, disminución de c-HDL), pero no un aumento significativo del grosor de la capa íntima-media de la arteria carótida44. McKimmie et al.45 estudiaron en pacientes diabéticos la relación entre el hígado graso (diagnosticado por TC) y el aumento de grosor de la capa íntima-media de la arteria carótida, llegando a la conclusión de que se trataba más de un epifenómeno que de una verdadera asociación. Sin embargo, Agarwal et al.46 evaluaron a 124 individuos con DM2 en tratamiento, de los cuales 71 (57,2%) mostraban NAFLD (mediante ecografía). Aquellos que presentaban NAFLD tuvieron una prevalencia más elevada de factores de riesgo cardiovascular (HTA, índice de masa corporal, obesidad abdominal, dislipidemia aterogénica) y un engrosamiento en la capa íntima-media arterial respecto a los que no presentaban hígado graso.
Análisis de asociación entre enfermedad por hígado graso no alcohólico, ateromatosis subclínica, episodios cardiovasculares y mortalidad
| Estudio | Población | Objetivo | Evaluación NAFLD | Prueba aterosclerosis subclínica | Morbilidad | Mortalidad | Conclusión |
| Mohammadi et al. (2011) | 84 pacientes NAFLD vs. 65 sujetos control | Asociación entre NAFLD y aterosclerosis subclínica | Ecografía abdominal | GIM y VMF | NAFLD y GIM, p = 0,001;NAFLD y VMF, p = 0,001 | La NAFLD (sin síndrome metabólico) está asociada a aterosclerosis subclínica | |
| Mohammadi et al. (2011) | 250 pacientes NAFLD vs. 85 sujetos control | Asociación entre NAFLD y aterosclerosis subclínica | Ecografía abdominal | GIM | NAFLD y GIM, p = 0,001 | La NAFLD (con síndrome metabólico) está asociada a aterosclerosis subclínica | |
| Poanta et al. (2011) | 56 pacientes DM2 (67%; 38/56 esteatosis vs. 33%; 18/56 sin esteatosis) | Prevalencia de NAFLD en DM2 y evaluar el estado de la arteria carótida | Ecografía abdominal | GIM | NAFLD y GIM, p = NS | La NAFLD se asocia a mayor número de factores de riesgo cardiovascular, pero no a aterosclerosis subclínica | |
| Agarwal et al. (2011) | 124 pacientes DM2 (57%; 71/124 NAFLD vs. 43%; 53/124 sin NAFLD) | Prevalencia de NAFLD en DM2 y correlación con enfermedad coronaria | Ecografía abdominal | GIM | NAFLD y GIM, p = 0,045 | La NAFLD se asocia a mayor número de factores de riesgo cardiovascular, así como a aterosclerosis subclínica | |
| McKimmie et al. (2008) | 623 pacientes DM2 (30%; 192/623 NAFLD) | Asociación entre NAFLD y aterosclerosis subclínica | Tomografía abdominal | GIM y TC coronaria | NAFLD y GIM, p = NS;NAFLD y TC, p = NS | La NAFLD es un estado inflamatorio y proaterogénico, pero es un epifenómeno respecto a enfermedad cardiovascular | |
| Lazo et al. (2011) | 11.371 pacientes (16,4%; 2.089 NAFLD; 3,1%; 426 NASH) | Evaluar las causas de mortalidad (cardiovascular, cáncer, hepática) en una población NAFLD | Ecografía abdominal | Respecto a sujetos sin esteatosis, NAFLD y NASH no se asociaron a mayor mortalidad (p = NS) | La NAFLD no fue relacionada con un incremento de mortalidad cardiovascular, hepática o por cáncer | ||
| Bhala et al. (2011) | 247 pacientes NAFLD vs. 264 pacientes VHC (todos Child-Pugh A y con fibrosis avanzada/cirrosis) | Evaluar y comparar las causas de morbilidad y mortalidad en ambas cohortes | Biopsia hepática | NAFLD vs. VHC: complicaciones hepáticas (p = 0,03); hepatocarcinoma (p = 0,03); episodios cardiovasculares (p = NS) | NAFLD vs VHC: mortalidad (p=ns) | La NAFLD tiene menos complicaciones hepáticas respecto a la hepatitis C, pero mortalidad similar | |
| Wong et al. (2011) | 612 pacientes con indicación clínica de coronariografía (58%; 356/612 NAFLD) | Asociación entre NAFLD y enfermedad coronaria, además mortalidad, IAM no mortal y revascularización durante el seguimiento | Ecografía abdominal | NAFLD 84,6% (301/356) vs. no NAFLD 64,1% (164/256) (p < 0,001) (presencia de enfermedad coronaria) | Desenlaces cardiovasculares: NAFLD 10% (30/301) vs. no NAFLD 11% (18/164) (p = NS) | La NAFLD se asocia a presencia de enfermedad coronaria, pero no con morbimortalidad en pacientes con enfermedad coronaria establecida |
DM2: diabetes mellitus tipo 2; GIM: grosor de la capa íntima-media de la arteria coronaria; IAM: infarto agudo de miocardio; NAFLD: enfermedad por hígado graso no alcohólico; NASH: esteatohepatitis no alcohólica; VHC: virus de la hepatitis C; VMF: vasodilatación mediada por flujo.
Los estudios acerca de la mortalidad cardiovascular son más escasos y menos clarificadores. Lazo et al. incluyeron a 11.371 pacientes en un estudio para evaluar la mortalidad por causa cardiovascular, hepática o por cáncer. Los pacientes se clasificaron como NAFLD cuando los individuos presentaban esteatosis hepática (mediante ecografía) y normalidad en las transaminasas (n = 2.089; 16,4%); NASH cuando presentaban esteatosis hepática y elevación en las transaminasas (n = 426; 3,1%). No se detectó un aumento de mortalidad relacionada con la presencia de NAFLD o NASH47. Bhala et al. compararon 247 pacientes con NAFLD y 264 pacientes con hepatitis C (que no habían recibido tratamiento previamente o no respondedores). Ambas cohortes eran Child-Pugh A y tenían fibrosis avanzada o cirrosis (evaluada mediante biopsia hepática). No se detectaron diferencias en términos de mortalidad global ni de accidentes cardiovasculares entre la cohorte NAFLD y hepatitis C, sin embargo sí hubo una menor tasa de complicaciones hepáticas (incluido hepatocarcinoma) en la cohorte NAFLD48. En un estudio reciente publicado por Wong et al.49 se incluyeron 612 pacientes con indicación clínica de coronariografía. Los pacientes se clasificaron en NAFLD (n = 356; 58,2%) mediante ecografía alterada o no NAFLD ecografía hepática normal. Se consideró enfermedad coronaria significativa una estenosis superior al 50% en, al menos, una arteria coronaria, lo que se detectó en 456 sujetos (76%). Los pacientes con hígado graso mostraron una prevalencia de enfermedad coronaria significativa del 84,6% (301/356), mientras que esta cifra descendía hasta el 64,1% (164/256) si no mostraban NAFLD (p < 0,001). No obstante, la NAFLD no pudo predecir la mortalidad cardiovascular en estos pacientes con enfermedad coronaria establecida.
ConclusionesLa NAFLD se relaciona estrechamente con la enfermedad cardiovascular, sobre todo con el engrosamiento de la capa íntima-media de la arteria carótida, como manifestación morfoestructural de la presencia de ateromatosis subclínica. No obstante, estos datos hay que interpretarlos con cierta cautela, ya que existe una gran heterogeneidad en los artículos publicados, por 3 motivos fundamentales: a) el diagnóstico de NAFLD se basa en la mayoría de los estudios en los hallazgos ecográficos (aun a pesar de que su seguridad diagnóstica es muy limitada ya que se trata de una técnica dependiente del explorador y solo permite la detección de esteatosis cuando esta es superior al 33% en la biopsia hepática), asociado o no a la alteración en los valores de las transaminasas, parámetro de seguridad diagnóstica también limitado; b) los criterios diagnósticos de aterosclerosis subclínica también son muy variables, lo que hace que haya que asumir en los diferentes análisis que los resultados patológicos de las distintas pruebas sean equivalentes, y c) no disponemos de estudios con suficiente potencia estadística que permitan confirmar si esta asociación entre NAFLD y episodios cardiovasculares se podría trasladar a la tasa de mortalidad.
En definitiva, esta asociación otorga una nueva dimensión a la práctica clínica diaria. La detección de un sujeto con NAFLD (enfermedad hepática más común en la población) debería alertarnos sobre la existencia de un mayor riesgo cardiovascular y ser más agresivos en la búsqueda de la prevención primaria, para lo cual resulta determinante la realización de pruebas de detección de aterosclerosis subclínica. El manejo correcto de la enfermedad por depósito graso no alcohólico, con la aparición de nuevas posibilidades terapéuticas, permitirá modificar la historia natural de la enfermedad tanto hepática como aterosclerótica.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
La investigación llevada a cabo para obtener estos resultados ha recibido financiación del Séptimo Programa Marco de la Comunidad Europea (FP7/2007-2013) bajo el acuerdo de subvención n° HEALTH-F2-2009-241762 para el proyecto FLIP.

