




El linfoma primario colorrectal comprende el 10-20% de los linfomas, y solo el 0,2-0,6% de las neoplasias malignas de intestino grueso. Su presentación clínica es inespecífica, y su diagnóstico tardío. La mayoría son linfomas no Hodgkin, siendo el subtipo histológico más frecuente el linfoma difuso de células B grandes1–3.
Presentamos el caso de un varón de 28 años, que acude a consulta por la aparición de una sensación de rigidez en fosa ilíaca derecha, que en menos de 30 días pasó a ser una masa de gran tamaño, objetivable a simple vista en decúbito supino. Unido a esto, presentaba anorexia marcada y pérdida de 9kg de peso de 3 meses de evolución, con sensación de plenitud tras la ingesta y dolor abdominal intermitente.
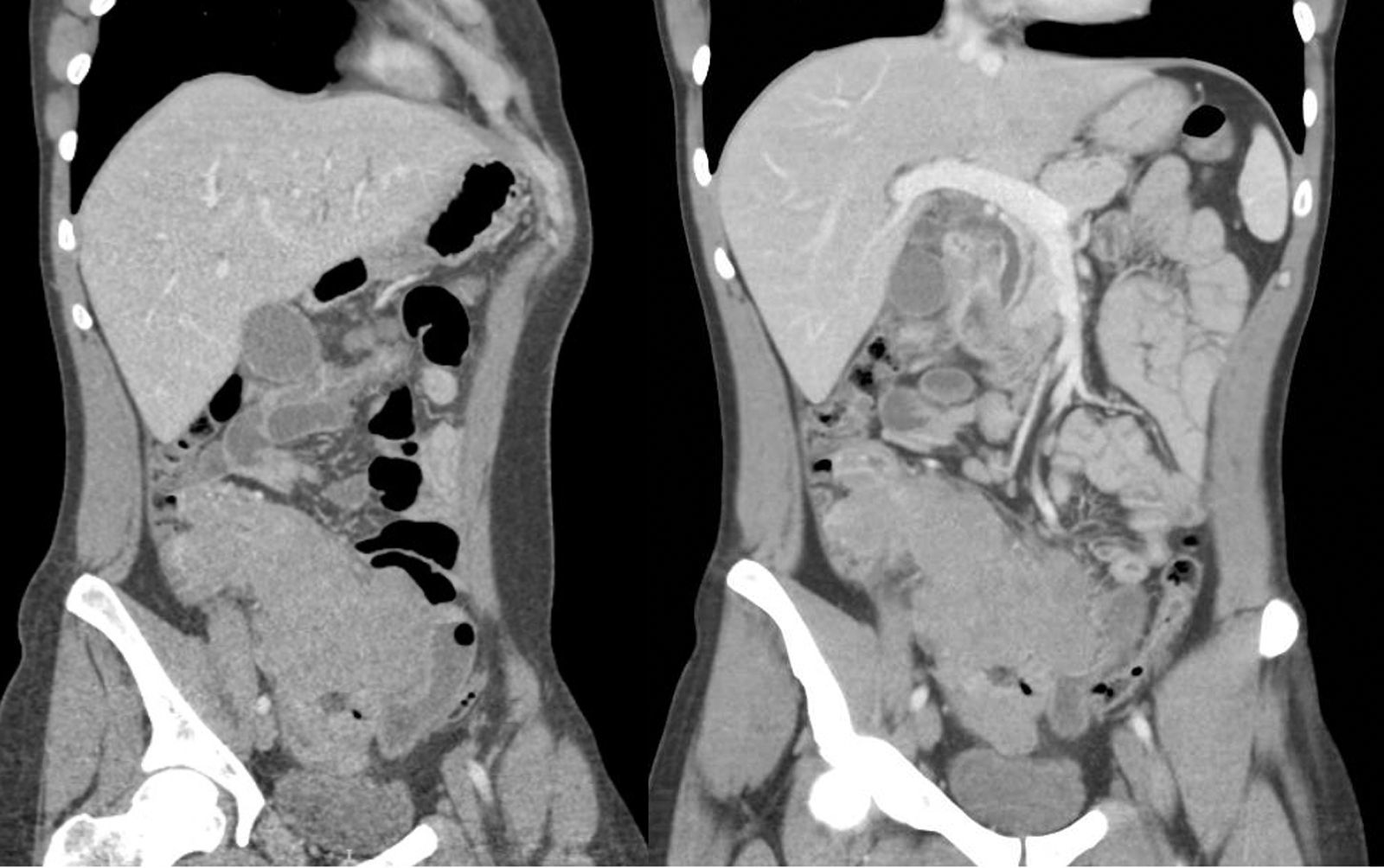
Se realizó una TAC abdominal con contraste, con hallazgo de engrosamiento marcado de las paredes del ciego y colon ascendente, adenopatías íleo-cólicas, engrosamiento de las paredes del duodeno, mínima dilatación de las vías biliares y lesiones hipodensas en el cuerpo-cuello pancreático (fig. 1).

Analíticamente destacaba hipertransaminasemia moderada y colestasis, con marcadores tumorales normales, incluido la ß2-microglobulina.
La colonoscopia mostraba una mucosa y calibre de colon normales hasta el colon ascendente donde no se conseguía buena distensión de la luz, y la ileoscopia amplias ulceraciones necróticas sugestivas de linfoma intestinal, que se biopsiaron. En la endoscopia oral se objetivaron ulceraciones de aspecto similar en duodeno proximal.
La histología fue informada como linfoma B difuso de célula grande, inmunofenotipo de centro germinal.
El PET-TAC de estadificación inicial mostró una extensa afectación linfomatosa intestinal en hipogastrio y fosa ilíaca derecha, con zona metabólica central compatible con necrosis, confirmándose una perforación intestinal posterior de colon ascendente mediante TAC, que requirió una hemicolectomía derecha urgente, con resección parcial de intestino delgado y anastomosis íleo-cólica latero-lateral, ante el hallazgo macroscópico de una gran masa tumoral que ocupaba todo el colon ascendente, ciego e íleon terminal, sin afectación de otros órganos vecinos.
La histología de la pieza quirúrgica puso de manifiesto un proceso linfoproliferativo de alto grado, de célula intermedia-grande, con áreas en cielo estrellado y focos de necrosis, asociado a virus de Epstein-Barr (EBER intensamente positivo), con alto índice proliferativo (Ki-67, prácticamente del 100%) y fenotipo centro germinal Bcl-2 negativo y CD20 positivo. Aunque la translocación t(8;14) fue negativa, se informó de linfoma de Burkitt asociado a virus de Epstein-Barr (VEB) en función de la celularidad, la asociación al virus de Epstein-Barr y el altísimo índice proliferativo del linfoma (Ki-67, prácticamente del 100%) típico de esta neoplasia frente a otros tipos de enfermedades linfoproliferativas. La pieza quirúrgica incluía ganglios linfáticos mesentéricos de gran tamaño, no infiltrados por el linfoma.
Se estadificó como un linfoma Burkitt estadio IVB y se inició quimioterapia en régimen de ensayo clínico. Tras 2 ciclos, presenta una excelente respuesta metabólica intermedia a la quimioterapia por PET-TAC, y está pendiente del último ciclo de quimioterapia.
El linfoma primario colorrectal es una neoplasia maligna muy infrecuente. El linfoma de Burkitt representa solo el 15% de todos los linfomas gastrointestinales4.
Los síntomas más frecuentes son dolor abdominal, anorexia y pérdida de peso1,3. La falta de especificidad implica, a menudo, un retraso en el diagnóstico.
La colonoscopia es una herramienta diagnóstica útil, a pesar del frecuente error de diagnóstico histológico de las biopsias, por la toma de muestras de tejido inespecífico adyacente. En la serie de Ding et al., las lesiones se identificaron macroscópicamente en un 95,7% de los casos (44/46), mientras que las biopsias fueron diagnósticas solo en el 21,7% (10/46)5. En el resto de casos, las biopsias no mostraron alteraciones o erraron en el diagnóstico, como ocurrió en nuestro caso.
Los hallazgos endoscópicos varían de ulceraciones a lesiones polipoideas2,3.
El linfoma de Burkitt se caracteriza por su asociación con la translocación cromosómica (8:14, 8:22 y 2:8), y la sobreexpresión del oncogén c-myc. Es el linfoma con mayor velocidad de crecimiento. Su incidencia es mayor en niños, y es infrecuente en la edad adulta.
Existen 3 variantes clínicas: endémico (asociado a la malaria y al VEB en la mayoría de los casos), asociado a inmunodepresión (relacionado con el VIH y con el VEB en un 40%) y esporádico, asociado en un 20% al VEB, como el nuestro. La influencia del VEB en la patogenia del linfoma de Burkitt se desconoce6.
El rápido crecimiento del tumor y el diagnóstico tardío (síntomas inespecíficos, aspecto macroscópico variable y baja rentabilidad de la biopsia endoscópica) implican frecuentemente la necesidad de una laparotomía urgente, antes de la confirmación histológica y del inicio del tratamiento, por la aparición de complicaciones (obstrucción intestinal, perforación o hemorragia).
El tratamiento de elección es la quimioterapia, agresiva en adultos por su peor pronóstico y respuesta al tratamiento7. La tasa de curación es del 90% en los países desarrollados. El diagnóstico precoz es fundamental para asegurar un tratamiento médico óptimo, y evitar las complicaciones de la cirugía.
FinanciaciónNo hemos recibido financiación para la realización del manuscrito.
Conflicto de interesesNinguno de los autores presenta conflicto de intereses.

