




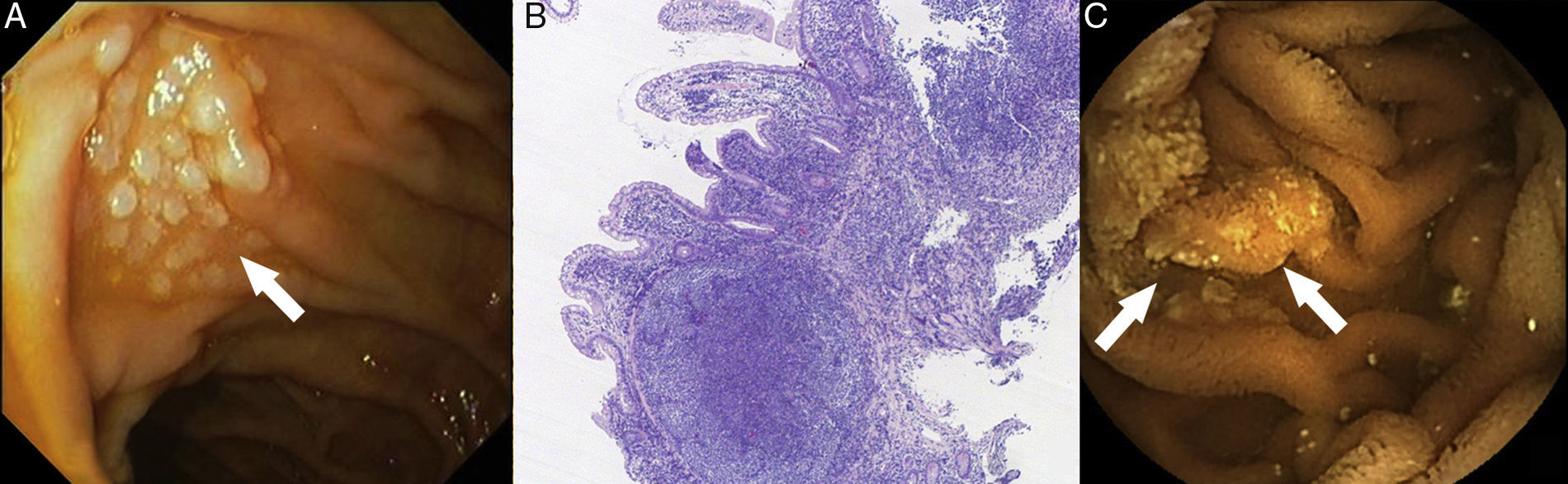
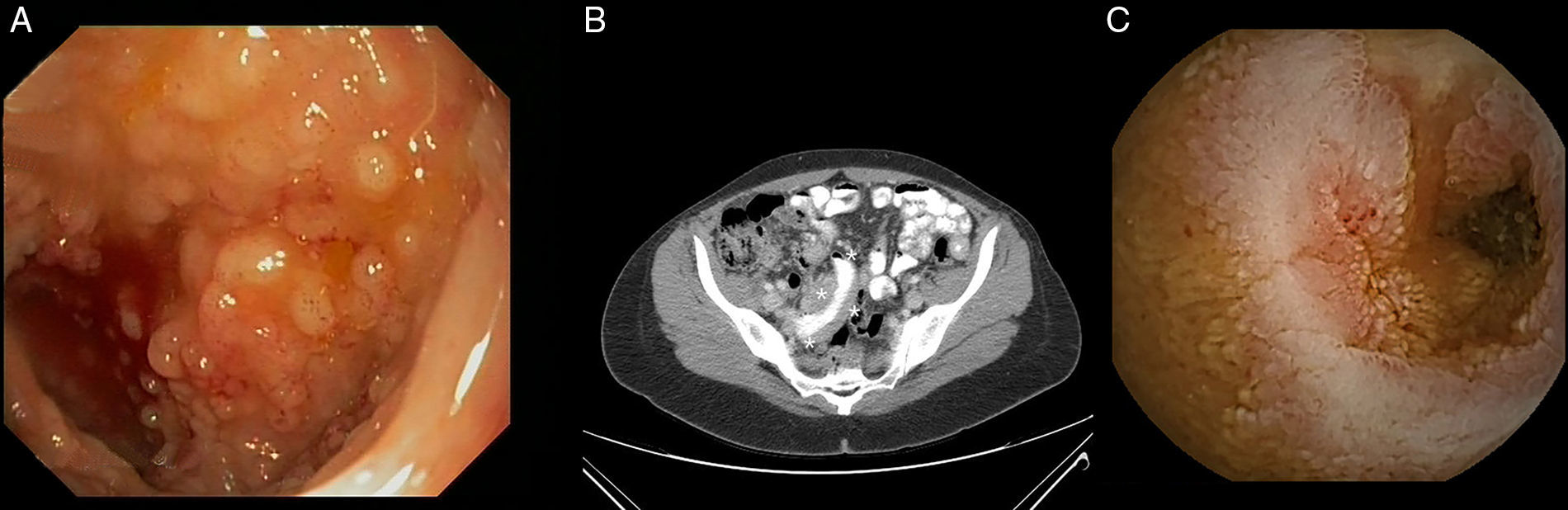
Los linfomas primarios gastrointestinales son neoplasias poco comunes, siendo la invasión gastrointestinal secundaria la causa más frecuente. El linfoma difuso de células grandes, el linfoma MALT y el linfoma del manto son los subtipos histológicos más frecuentes. La Organización Mundial de la Salud reconoce desde 2008 un nuevo subtipo de linfoma, el linfoma folicular primario intestinal (LF-PI). Presentamos el caso de 2 pacientes diagnosticados en nuestro centro de un LF-PI. El primer caso es el de una mujer de 56 años portadora de una mutación en el exón 12 del gen HMSH-2, descubierta tras una cirugía (histerectomía con doble anexectomía) por un adenocarcinoma de ovario 12 años atrás (T1N0M0; clasificación TNM, 7.ª edición). Hace 3 años y de forma incidental, fue diagnosticada de un LF-PI de duodeno tras detectarse durante una gastroscopia rutinaria una lesión blanquecina, nodular y elevada en vecindad con la papila de Váter (fig. 1A). Las biopsias demostraron la presencia en la lámina propia de una proliferación linfoide de células B, formada por linfocitos pequeños e irregulares, y con algún centroblasto aislado. El estudio inmunohistoquímico fue positivo para los marcadores CD20, CD10, BCL-2, BCL-6 y negativo para CD3, CD5 y ciclina-D1 (fig. 1B). El estudio de extensión (TAC toraco-abdominal, biopsia de médula ósea y PET) descartó enfermedad a distancia, si bien se identificaron 3 lesiones más, de similares características, en yeyuno medio, distal e íleon terminal, durante la enteroscopia con cápsula (EC) (fig. 1C). Tras 4 ciclos de tratamiento con rituximab la paciente alcanzó la remisión completa. Pasados 2 años y en un nuevo control se evidenció recurrencia endoscópica e histológica, pero sin progresión radiológica. Ante la negativa de la paciente a recibir nuevo tratamiento, se decidió tratamiento conservador. Pasados 12 meses, la paciente permanece estable y sin evidencia de progresión tumoral. El segundo caso presentado hace referencia a una mujer de 48 años estudiada por astenia de 6 meses de evolución, vómitos y dolor abdominal relacionado con la menstruación. Se realizó estudio endoscópico convencional (gastroscopia e íleo-colonoscopia) detectándose en íleon terminal unas lesiones mucosas polipoides compatibles con hiperplasia folicular linfoide (fig. 2A). Dada la persistencia de los síntomas, se indicó una TAC abdominal donde se evidenció un engrosamiento mural en un segmento corto de íleon terminal junto con adenopatías regionales y retroperitoneales, sin otros hallazgos relevantes (fig. 2B). La EC confirmó los hallazgos radiológicos y detectó una estenosis mucosa de novo, infranqueable para la cápsula (fig. 2C). Tras 2 semanas de tratamiento con budesonida (9mg/24h) el dispositivo fue expulsado por vía natural, sin incidencias. Se revisaron nuevamente las biopsias obtenidas previamente, observándose en esta ocasión grupos celulares con un inmunofenotipo compatible con un LF-PI de íleon. Se indicó tratamiento quimioterápico con CHOP+rituximab, observándose tras 2 ciclos de tratamiento mejoría radiológica en la TAC de control.
 Lesión sobreelevada blanquecina y nodular yuxta-papilar, con (B) proliferación de células B formada por centrocitos y centroblastos (H&E ×40). (C) LF-PI duodenal mediante enteroscopia con cápsula.")
 Lesiones polipoides en íleon terminal, con (B) engrosamiento mural del íleon en la TAC. (C) Estenosis ileal ulcerada e infranqueable durante la enteroscopia con cápsula.")
El LF-PI es una variante del LF ganglionar1. Se caracteriza por ser un linfoma poco común, representando el 1 al 3,6% de todos los linfomas primarios gastrointestinales2. La prevalencia suele ser mayor entre las mujeres de mediana edad. Con frecuencia son asintomáticos (43%), siendo el dolor abdominal (28%), las náuseas-vómitos (8%) y el sangrado digestivo (6%) los síntomas más comunes cuando estos están presentes3. Generalmente son tumores unifocales, siendo la segunda porción del duodeno en vecindad con la ampolla de Váter la localización más frecuente. Sin embargo, al igual que en nuestra primera paciente, se han descrito casos de LF-PI multifocales. En estos, la EC puede ser de gran utilidad para la detección y localización de todas las lesiones sincrónicas, al tratarse de una técnica inocua y bien tolerada. Así mismo, también puede servir de guía para la toma de biopsias cuando la endoscopia convencional resulta negativa4. Endoscópicamente, el LF-PI suele presentarse como pequeñas lesiones polipoides recubiertas por mucosa normal. Sin embargo, en raras ocasiones se presenta en forma de úlceras que pueden estenosar la luz intestinal5,6. Histológicamente, presentan un patrón de crecimiento nodular compuesto por centros germinales formados por centrocitos y centroblastos con un inmunofenotipo típico positivo para los marcadores CD19, CD20, CD22 y CD79a y negativo para CD43, CD5 y ciclina-D17. El estudio de extensión debe incluir estudio endoscópico, una TAC toraco-abdominal y/o PET y un estudio de médula ósea. La clasificación de Ann-Arbor, así como la de Lugano resultan útiles para la estadificación tumoral, en especial esta última cuando LF-PI es multifocal8. Actualmente se desconoce cuál es la estrategia de tratamiento óptima, debiendo personalizarse esta según las características particulares de cada paciente. En pacientes asintomáticos y estadios localizados de la enfermedad, el manejo puede ser expectante. Sin embargo, el tratamiento debe iniciarse cuando los pacientes presentan síntomas y/o progresión de la misma, variando este según el grado histológico y clínico. La radioterapia suele ser el tratamiento de elección en aquellos casos en los que la enfermedad está localizada y no existen factores de mal pronóstico, reservándose la monoterapia con rituximab (Ac monoclonal anti-CD20) para aquellos casos seleccionados en los que se desea a evitar los efectos indeseados de la radioterapia. Si por el contrario la enfermedad está diseminada o presenta factores de mal pronóstico, está indicado el tratamiento sistémico con quimioterapia (CHOP, CVP) combinado a no con rituximab9.

