





La identificación del gen HFE implicado en la mayoría de casos de hemocromatosis hereditaria ha constituido un gran avance para el diagnóstico de esta entidad; sin embargo, hay otras formas de hemocromatosis hereditarias no relacionadas con dicho gen. En los últimos años se ha avanzado en el conocimiento de la interrelación entre las manifestaciones fenotípicas de diferentes tipos de hemocromatosis y las mutaciones en 4 genes, algunos de ellos recientemente identificados (HFE, TfR2, HAMP y HJV). No obstante, la división de la hemocromatosis en 4 tipos (en la edad adulta se presentan las relacionadas con mutaciones HFE o TfR2, y en la juventud las HJV o HAMP), basada exclusivamente en criterios genéticos, puede no ser exacta pues probablemente aparecerán nuevos genes implicados, y las diferentes asociaciones de mutaciones entre estos genes también influyen en el fenotipo (herencia digénica), sin olvidar los diferentes factores ambientales del huésped. Por ello, todas estas variantes de hemocromatosis deberíamos englobarlas dentro de un único síndrome clinicopatológico. Dentro de los avances acerca de la patogenia de la hemocromatosis hereditaria ligada al gen HFE resalta la nueva hipótesis, en la que se implica muy sugestivamente a la hepcidina.
CONSIDERACIONES FISIOLOGICAS ACERCA DEL TRANSPORTE Y LA ABSORCION INTESTINAL DEL HIERRO
La ingesta habitual de hierro en la dieta occidental es de 10-30 mg diarios, y las personas normales absorben sólo 1 mg. Los pacientes con hemocromatosis pueden absorber 3-6 mg de hierro al día; se acumula en el organismo a razón de un ritmo aproximado de 0,5 g/año, y llega en estadios avanzados a sobrecargas de hierro superiores a 20 g (valores normales hasta 4-5 g).
Mientras que el hierro es esencial para el crecimiento y el metabolismo celular, al contener electrones impares puede desempeñar un papel decisivo en la generación de radicales libres de oxígeno, requiriéndose sistemas que ajusten la concentración de hierro intracelular a valores adecuados para mantener sus necesidades metabólicas, pero sin alcanzar concentraciones tóxicas. El hierro también es requerido para el crecimiento de microorganismos, por lo que es importante su control para mantener la inmunidad del huésped frente a las infecciones bacterianas1. Los linfocitos intraepiteliales del intestino delgado producen factores de necrosis tumoral alfa (TNF-*) en respuesta al hierro de la dieta2. La ingesta de hierro produce una señal a través de HFE a los linfocitos intraepiteliales, estimulando la producción de TNF-*, con la consiguiente disminución de la absorción de hierro por los enterocitos.
El hierro de la dieta está en forma de hierro-hem y de hierro no-hem3. El hierro, al ser un metal hidrofílico, es incapaz de atravesar las membranas celulares, requiriéndose sistemas especializados de transporte tanto para mantener al hierro en un estado soluble apto para circular en la corriente sanguínea como para poder atravesar las membranas celulares.
El hierro-hem proveniente de la hemoglobina o de la mioglobina se absorbe mejor que el hierro no-hem, aunque su mecanismo de absorción aún está por aclarar, y puede haber un receptor hem en el reborde en cepillo de las vellosidades. Algunos alimentos, como carnes y pescados, contienen el 30-60% en forma de hierro-hem; por el contrario, los vegetales, los cereales y las frutas contienen sólo hierro no-hem, la mayoría en forma de sales férricas.
El hierro férrico (Fe3) es relativamente insoluble en agua cuando el pH luminal es superior a 3. Por el contrario, el hierro ferroso (Fe2) permanece soluble incluso con un pH de 8. Varias substancias de bajo peso molecular (azúcares, aminoácidos, citratos, ascorbatos) quelan el hierro intraluminal, aumentando su solubilidad y facilitan su absorción; pero otros agentes (carbonatos, oxalatos, fosfatos, tanatos y fitatos vegetales) forman complejos insolubles con el hierro e inhiben su absorción4.
El hierro absorbido se encuentra en 3 compartimentos: a) el incorporado a la hemoglobina (dos tercios del hierro corporal está incorporado en la hemoglobina); b) el hierro plasmático transportado por la transferrina (Tf), y c) el hierro de los depósitos ligado a proteínas, como la ferritina y la hemosiderina. Cada molécula de Tf puede acoplar dos moléculas de Fe3 (transferrina diférrica); normalmente está ocupado el 30% de los lugares de acoplamiento de la Tf. La ferritina es una molécula compleja que alberga hierro soluble, movilizable y en forma atóxica; una envoltura de ferritina puede acomodar 4.500 átomos de hierro. La hemosiderina está constituida por unidades de ferritina y otros radicales. Debido a que el eritrón requiere 20 mg de hierro diariamente, y a que sólo entran en el organismo 1-2 mg al día a través del intestino, el hierro se recicla continuamente mediante la destrucción de los glóbulos rojos en los macrófagos, seguido de una incorporación del hierro a la Tf sérica para que se deposite en la médula ósea y se reincorpore a los glóbulos rojos. Una tercera parte del hierro almacenado se encuentra normalmente en el hígado, sobre todo en forma de ferritina.
Se han descubierto dos tipos de receptores de la Tf: el TfR1 y el TfR2.
La producción de TfR1 se regula por las denominadas proteínas reguladoras del hierro (iron regulatory proteins, IRP); dichas proteínas se unen al elemento respondedor del hierro (iron responsive element, IRE) situado en una región específica del ARNm de los TfR15 y en la ferritina. En deficiencias de hierro, el IRP se une al IRE incrementándose la cantidad del TfR1 en la membrana celular y disminuyendo la cantidad de ferritina intracelular, con lo que aumenta la captación de hierro y disminuye su almacenamiento; lo contrario ocurre en condiciones de exceso de hierro.
Los TfR2 se expresan principalmente en hepatocitos, aunque también en las criptas duodenales y las células eritroides; tienen una menor capacidad para acoplar hierro que los TfR1 (unas 25-30 veces menor)6. El HFE y el TfR2 se disponen juntos en las células de las criptas, pero la distribución del TfR2 en las criptas es diferente del TfR1. El ARNm del TfR2 se expresa a muy bajos valores en el duodeno, e in vitro7. Mutaciones del TfR2 pueden dar lugar a la hemocromatosis tipo 3.
Recientemente se ha descubierto que el transportador de hierro DMT-1 (divalent metal transporter-1, antes DCT-1 o divalent cation transporter-1, Nramp2 o natural resistance of macrophages associated protein-2) es el más importante sistema de captación de hierro independiente de la Tf en los enterocitos, y también en las vesículas endosómicas en diferentes tejidos; hay 4 formas isomorfas, dos IRE+vas y dos IRE-vas8.
En el duodeno varias proteínas facilitan la captación y exportación de hierro, como la Dcytb, la DMT-1, la hephaestina y la ferroportina 1. En la tabla I se mencionan las proteínas, transportadores y reguladores implicados en el metabolismo del hierro.
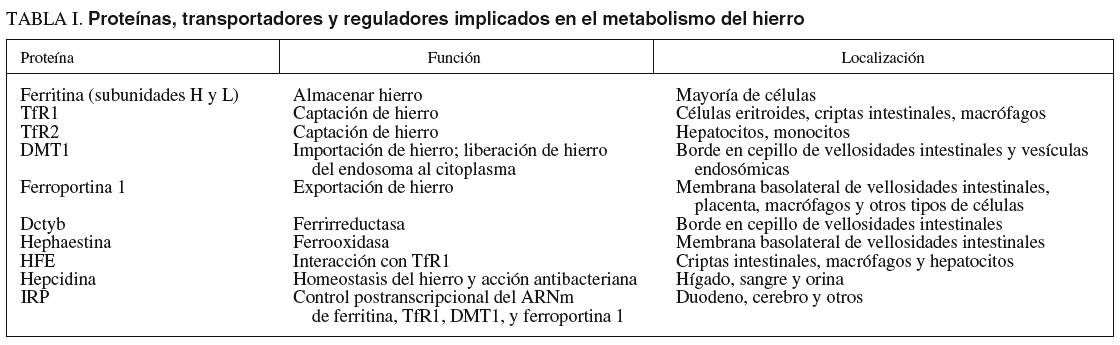
En la luz intestinal el hierro de la dieta está predominantemente en forma de Fe3, reducido a Fe2 por la ferrirreductasa del reborde en cepillo de los enterocitos denominada Dcytb o citocromo b9, siendo ulteriormente bombeado por los DMT-1 localizados en la membrana apical de las vellosidades al interior del enterocito (fig. 1)10. El resto del hierro de la dieta está en forma predominantemente de hierro-hem, que es captado por un mecanismo endocítico diferente11, puede que mediante un receptor hem en el reborde en cepillo de las vellosidades, y una vez en el interior del enterocito el hierro sea liberado del hem mediante una hem-oxigenasa y ulteriormente almacenado o transferido fuera del enterocito por un mecanismo similar al del Fe2.
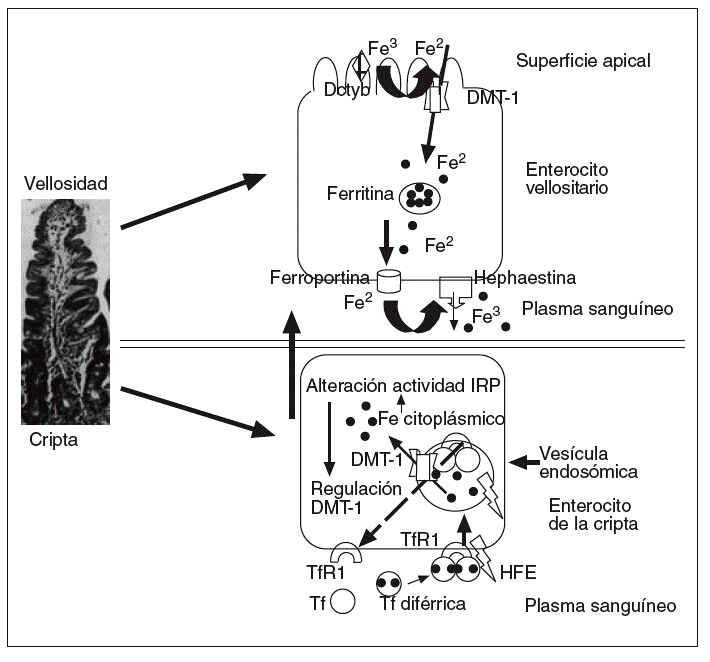
Fig. 1. En la vellosidad intestinal, el Fe3 proveniente de la dieta es primero reducido a Fe2 por la ferrirreductasa Dctyb, tras lo cual es vehiculado por el transportador denominado DMT-1 situado en la porción apical del enterocito al citoplasma celular, y ulteriormente vertido a la circulación mediante primero un transportador denominado hephaestina, que es una ferrooxidasa homóloga de la ceruloplasmina situada en la membrana basolateral de los enterocitos, catalizando la oxidación del Fe2 a Fe3, y ulteriormente por la ferroportina 1, que es un exportador de hierro, también expresado en la membrana basolateral de los enterocitos, estando pues la salida del hierro mediada por la ferroportina 1 en conjunción con la hephaestina. En la cripta intestinal, la transferrina diférrica, constituida por dos moléculas de Fe3 vehiculadas en el plasma por la transferrina (Tf), se acopla al receptor de la transferrina (TfR1) y a la proteína HFE situados en la membrana basolateral de las células de las criptas intestinales, incorporándose al interior en forma de vesículas endosómicas. Al disminir el pH el Fe3 es liberado del complejo TfR1-Tf y reducido a Fe2, saliendo al citoplasma por medio del transportador de hierro DMT-1 situado en la membrana endosómica. El valor de Fe2 citoplásmico actúa a través del acoplamiento de IRP a IRE sobre la producción del ARNm del transportador DMT-1. El Fe2 está disponible para los requerimientos metabólicos o para almacenarse en forma de ferritina. El TfR1 con la Tf son reciclados a la membrana plasmática donde las moléculas de Tf son liberadas a la circulación.
La salida del Fe2 desde el citoplasma a través de la membrana basolateral del enterocito a la sangre está mediada por la hephaestina en conjunción con la ferroportina 1. La hephaestina es una ferroxidasa (homóloga de la ceruloplasmina) situada en la membrana basolateral de los enterocitos duodenales que facilita la exportación de hierro desde el enterocito a la sangre, catalizando la oxidación del Fe2 a Fe3 12. Mediante la ferroportina 1 (FP1, IREG1 o iron regulated protein 1, MTP1 o metal transport protein), que es una exportadora de hierro, expresada también en la membrana basolateral de los enterocitos duo-denales, se completa la transferencia del Fe3 a la circulación13 (fig. 1). Mutaciones de la ferroportina 1 se pueden asociar a la rara forma de hemocromatosis tipo 4.
En la cripta intestinal, la transferrina diférrica constituida por dos moléculas de Fe3 vehiculadas en el plasma por la Tf, se acopla al TfR1 y al HFE, situados en la membrana basolateral de las células de las criptas intestinales, incorporándose al interior en forma de vesículas endosómicas14. Con un pH inferior intracelular, el Fe3 es liberado del complejo TfR1-Tf y reducido a Fe2, saliendo al citoplasma por medio del transportador de hierro DMT-1 situado en la membrana endosómica. La concentración de Fe2 citoplásmico actúa a través del acoplamiento de IRP a IRE sobre la producción del ARNm del transportador DMT-1. El Fe2 está disponible para los requerimientos metabólicos o para almacenarse en forma de ferritina. El HFE y el TfR1 con la Tf son reciclados a la membrana plasmática, desde donde las moléculas de Tf son liberadas a la circulación (fig. 1).
Debido a que la absorción de hierro se da primordialmente en las vellosidades del duodeno y yeyuno, la localización del HFE en el citoplasma y la membrana basolateral de los enterocitos de las criptas sugiere que esta proteína no transportadora, ni acopladora del hierro, está involucrada en predeterminar la capacidad absortiva intestinal del hierro por mecanismos inciertos, antes de que dichas células emigren a las vellosidades15,16 (fig. 1). Se ha detectado HFE en casi todos los tejidos salvo en el cerebro y en los linfocitos, que es más abundante en el hígado, sobre todo en los hepatocitos (y a una concentración 10 veces inferior en las células de Kupffer)17, detectándose en el intestino delgado en las células de las criptas, pero no en los enterocitos maduros de las vellosidades. Se postula que la función normal del gen HFE es promover la captación del hierro mediado por los TfR1 en las células de las criptas del intestino delgado15. El TfR1 no participa en la captación del hierro luminal intestinal, sino que, situado en la membrana basolateral, está más implicado en la transferencia de hierro del enterocito de la cripta al plasma, y viceversa. La proteína HFE se acopla a los TfR1, pero es controvertido si lo hace con los TfR28,18. La HFE y el TfR2 no contienen IRE, por lo que su expresión no está regulada por el hierro19.
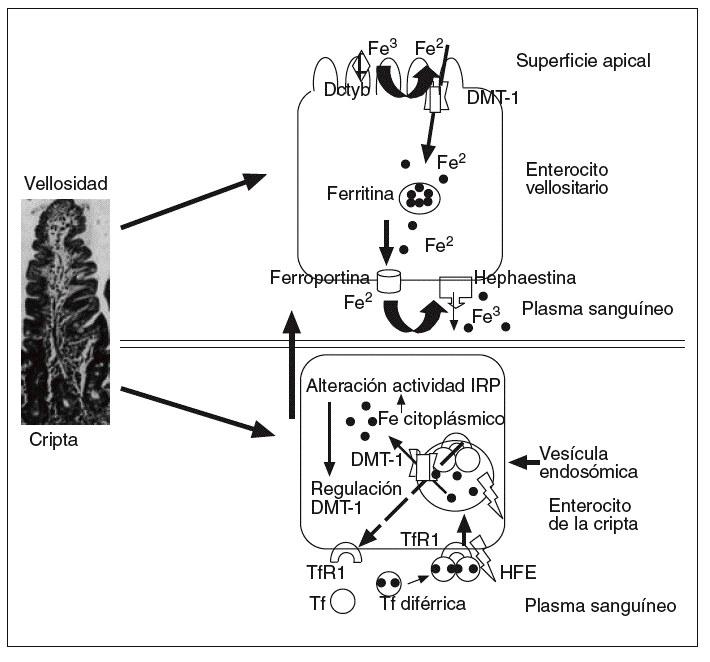
La captación del hierro ligado a la Tf en el hígado se da en parte mediante los TfR1 y también por vías independientes de los TfR1. En el hígado, el TfR2 se expresa en abundancia7 y está probablemente implicado en la captación de hierro por un mecanismo independiente del TfR1; la expresión del TfR1 es poco numerosa y condicionada por la sobrecarga de hierro, y puede estar ausente en pacientes con hemocromatosis, por lo que parece improbable que contribuya significativamente a la captación de hierro en el hígado20. La captación del hierro no ligado a la Tf en el hígado puede estar vinculada al estimulador del transporte de hierro (SFT)21 o al DMT-1. La expresión del transportador DMT-1 aumenta en los hepatocitos en relación con la carga de hierro, por lo que puede actuar facilitando la entrada de hierro a través de la membrana hepatocitaria22. La ferroportina 1 permite la salida del hierro a través de la membrana del hepatocito13; después el Fe2 será oxidado por la ceruloplasmina y acoplado a la transferrina en el plasma23 (Tf diférrica) (fig. 2). Como mencionamos con anterioridad, el HFE se expresa predominantemente en los hepatocitos y a una concentración muy inferior en las células de Kupffer.
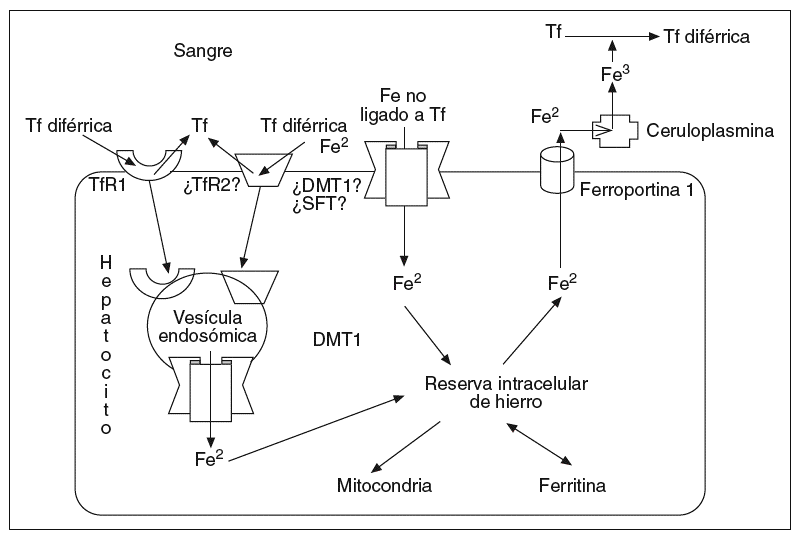
Fig. 2. El TfR2 se expresa abundantemente en el hígado y probablemente esté implicado en la captación de hierro por un mecanismo independiente del TfR1; la expresión del TfR1 está limitada por la sobrecarga de hierro, llegando a estar ausente en pacientes con hemocromatosis, por lo que parece improbable que contribuya significativamente a la captación de hierro en el hígado. La captación del hierro no ligado a la Tf en el hígado puede estar vinculada al estimulador del transporte de hierro (SFT), o al DMT-1. La expresión del transportador DMT-1 aumenta en los hepatocitos en relación con la carga de hierro, por lo que puede actuar facilitando la entrada de hierro a través de la membrana hepatocitaria. La ferroportina 1 parece permitir la salida del hierro a través de la membrana hepatocitaria; posteriormente, el hierro será oxidado a Fe3 por la ceruloplasmina y acoplado a la Tf en el plasma.
HEMOCROMATOSIS HEREDITARIAS
Antes de describir los tipos de hemocromatosis conviene recordar las diferentes entidades clínicas que pueden cursar con sobrecarga de hierro (tabla II).

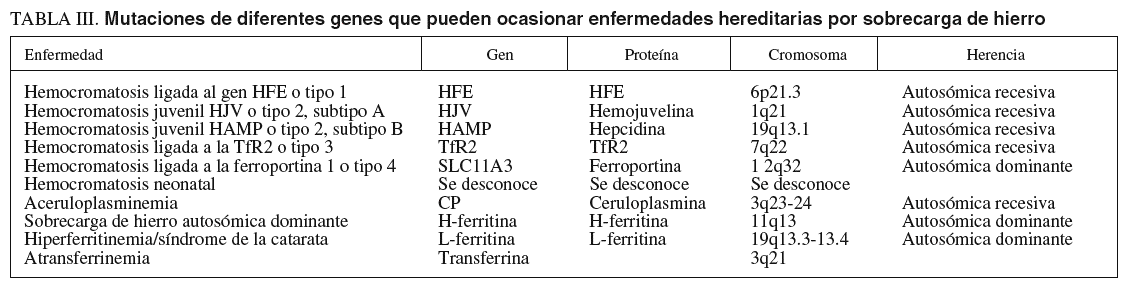
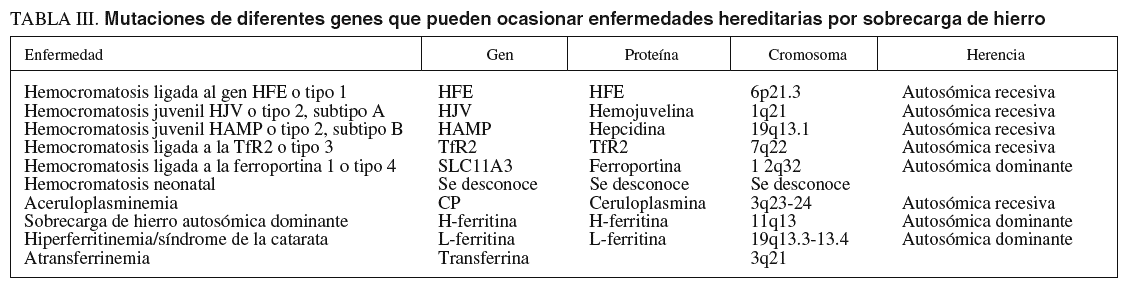
Los síntomas clásicos diabetes, pigmentación bronceada de la piel y cirrosis fueron descritos por primera vez en 1865 por Trousseau24, empleándose el término hemocromatosis por von Recklinghaussen25. En 1935, Sheldon comprobó que era una enfermedad hereditaria, causada por excesivos depósitos de hierro en los tejidos26. Saddi27, en 1974, demostró que era una enfermedad autosómica recesiva. Simon comprobó que estaba ligada al brazo corto del cromosoma 6 codificando un complejo mayor de histocompatibilidad de clase I, el HLA-A328. En 1996 el gen HFE fue finalmente identificado29. Ulteriormente, se han detectado otras formas de hemocromatosis hereditarias (tabla III).
Es importante recalcar que para el diagnóstico de hemocromatosis no basta con la detección de las mutaciones genéticas, sino que deben coexistir manifestaciones fenotípicas de la enfermedad.
La base de datos de la Online Mendelian Inheritance in Man (OMIM)30, describe 4 tipos de hemocromatosis hereditaria causadas por mutaciones en diferentes genes.
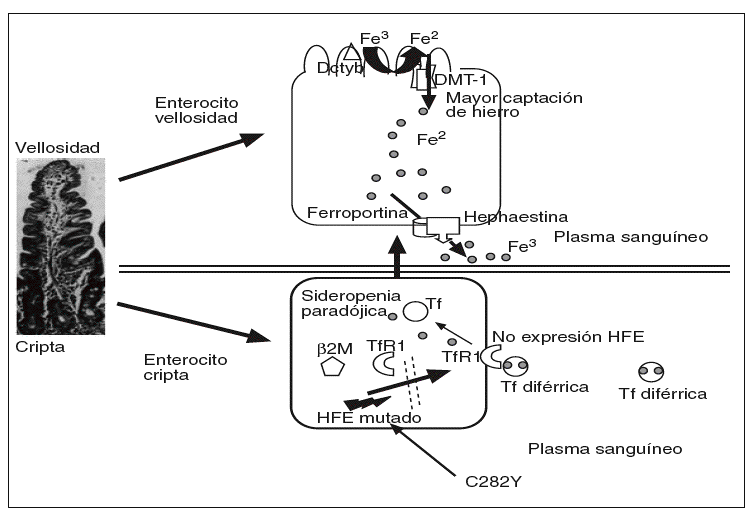
En la hemocromatosis ligada al gen HFE, o tipo 1, u OMIM 235200, se desconoce con exactitud el mecanismo del fallo en la regulación de la absorción del hierro. Suele manifestarse en la cuarta o quinta década de la vida, se transmite con carácter autosómico recesivo y responde muy bien a las flebotomías. La mutación C282Y, al impedir la unión de la beta-2 microglobulina al HFE, éste no puede ser transferido a la membrana plasmática basolateral y se queda en el citosol29, y puede impedir la captación del hierro circulante mediada por los TfR1 en las células de las criptas31, y así dar una falsa señal de que los depósitos de hierro están bajos (sideropenia paradójica); como resultado de los bajos valores de hierro en los enterocitos de las criptas, se incremetaría la producción de DMT-1 en los enterocitos maduros de las vellosidades y, por tanto, captarían más hierro (fig. 3).
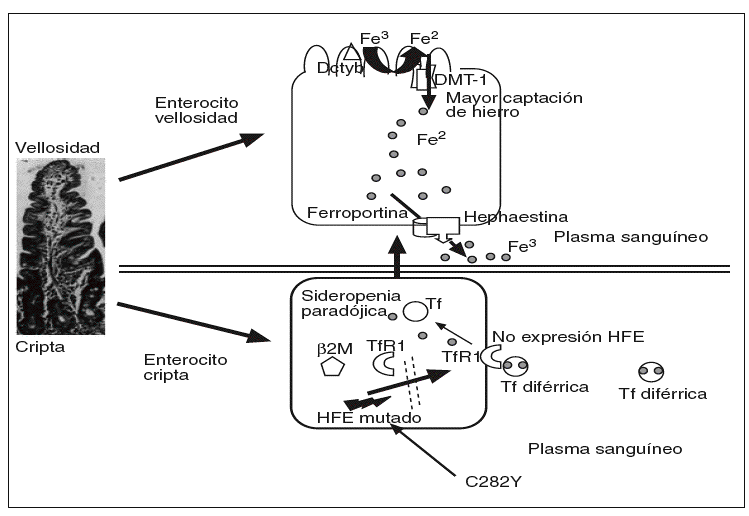
Fig. 3. En la mutación C282Y, al impedir la unión de laß2-microglobulina a la proteína HFE, ésta no puede ser transferida a la membrana basolateral de las células de las criptas, por lo que permanece en el citosol, lo que impide la captación de hierro mediada por el TfR, y así proporciona una falsa señal de que los depósitos de hierro en el enterocito de la cripta están disminuidos (sideropenia paradójica); como resultado de las bajas concentraciones de hierro intracelular, los enterocitos emigrando a lo alto de las vellosidades, aumentarían la producción de DMT-1 en los enterocitos maduros y, por tanto, captarían más hierro en pacientes con hemocromatosis ligada al gen HFE.
La mutación C282Y con carácter homocigoto es la más prevalente en los países de origen céltico o nórdico europeo, pues se da en 5 de cada 1.000 personas de este origen, haciendo que esta prevalencia sea 4 veces superior al déficit de alfa-1 antitripsina, 10 veces al de la fibrosis quística, 25 veces al de la fenilcetonuria y 75 veces al de la enfermedad de Wilson32. En Australia, un 85-90% de hemocromatósicos presentan homocigosis para la mutación C282Y. Esta mutación es rara en África, Polinesia, Asia, así como en los aborígenes australianos32.
También los que presentan la mutación C282Y/H63D pueden padecer formas más leves de hemocromatosis33. La mutación H63D en la homocigosis es poco penetrante, y se ha relacionado con algún caso aislado de hemocromatosis leve34. La S65C también es poco penetrante35. Otras formas con doble heterocigosis, como la C282Y/ S65C o la H63D/S65C permanecen aún controvertidas respecto a si inducen manifestaciones fenotípicas.
Wallace36 describe una nueva mutación del HFE, la IVS3 + 4T > 5 en un paciente con hemocromatosis, en heterocigosis con la C282Y. Piperno37 describe dos nuevas mutaciones antisentido del HFE, la G168T y la G169A en 5 pacientes italianos que asociadas a la C282Y dan lugar a una expresión fenotípica de la hemocromatosis.
En el futuro, el cribado para la mutación C282Y puede tener otras ventajas diagnósticas, puesto que aunque dicha mutación no es un factor de alto riesgo para la enfermedad cardiovascular o para el Alzheimer, sí se ha objetivado que en las mujeres fumadoras o hipertensas incrementa el riesgo de enfermedad cardiovascular38. También el riesgo de infarto de miocardio y de arteriosclerosis se ha relacionado con los depósitos de hierro39. Además, el riesgo de Alzheimer aumenta cuando la mutación C282Y se asocia con la apolipoproteína E4, y más aún en presencia del alelo C2 de la Tf40.
También se ha detectado que la hemocromatosis puede provenir de una herencia digénica de mutaciones combinadas del HFE, TfR2, HJV o HAMP, y la gravedad de la enfermedad se correlaciona sobre todo con mutaciones HAMP41 (v. más adelante, respecto a la hemocromatosis juvenil).
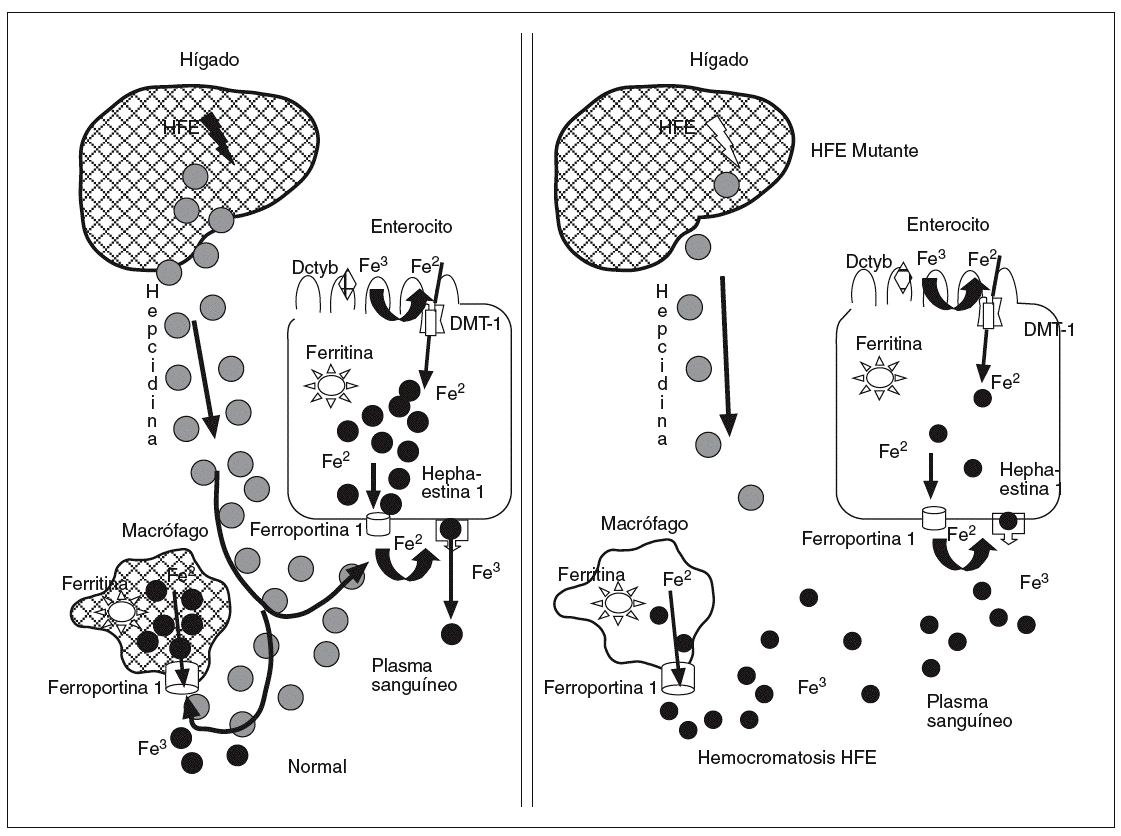
La hepcidina es un péptido de 25 aminoácidos sintetizada por los hepatocitos, que desempeña un papel clave como sensor de los depósitos de hierro, transmitiendo esta información al epitelio intestinal y a los macrófagos, donde por mediación de la ferroportina 1 permite una mayor o menor salida de hierro a la sangre, dependiendo de que los niveles de hepcidina en sangre estén disminuidos o elevados42,43. Es probable que el HFE tenga un papel en la modulación de la hepcidina, de forma que las mutaciones del HFE pueden disminuir la expresión de la hepcidina, lo que se plasmaría en una incontrolada liberación de hierro por enterocitos y macrófagos. Por ello, en una nueva hipótesis sobre la patogenia de la hemocromatosis hereditaria ligada al HFE se implica muy sugestivamente a la hepcidina44 (fig. 4). La hepcidina se correlaciona inversamente con los valores duodenales de Dcytb, DMT-1 y ferroportina 144. La expresión de la hepcidina está claramente modulada por la interleucina-645. En ratones y humanos se demuestra que la interleucina-6 es capaz de inducir la expresión de hepcidina y disminuir significativamente las concentraciones de hierro sérico. También previamente se objetivó que la hepcidina ejerce un papel significativo en la actividad antibacteriana45,46 y en la anemia crónica inflamatoria.
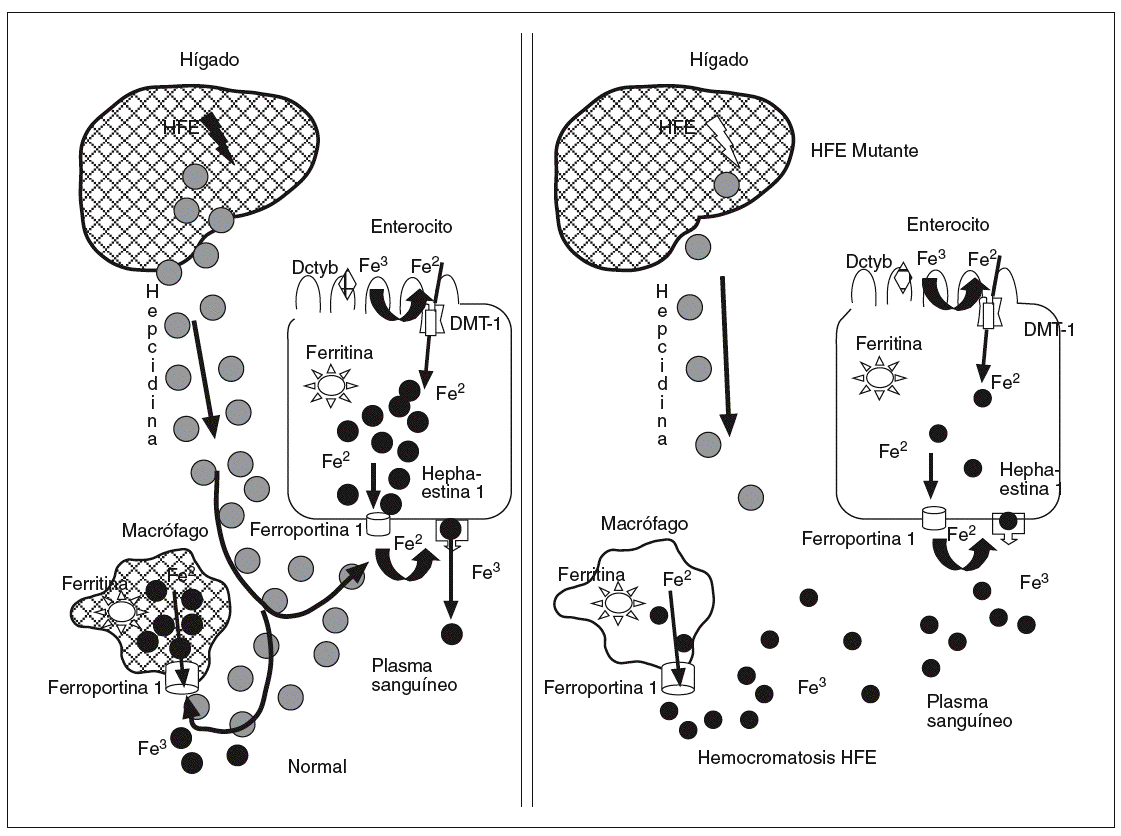
Fig. 4. La hepcidina es un péptido sintetizado por los hepatocitos, que desempeña un papel clave como sensor de los depósitos de hierro, transmitiendo esta información al epitelio intestinal y a los macrófagos, donde por mediación de la ferroportina 1 permite una mayor o menor salida de hierro a la sangre en función de que los valores de hepcidina estén disminuidos o elevados. Es probable que el HFE tenga un papel en la modulación de la hepcidina, por lo que las mutaciones del HFE pueden disminuir la expresión de la hepcidina, lo cual se plasmaría en una incontrolada liberación de hierro por enterocitos y macrófagos. Por ello, en una nueva hipótesis sobre la patogenia de la hemocromatosis hereditaria ligada al HFE se implica muy sugestivamente a la hepcidina.
La hemocromatosis juvenil, o tipo 2, es una forma más grave de enfermedad, muy poco frecuente, de carácter autosómica recesiva, que afecta por igual a varones y mujeres, soliendo manifestarse clínicamente antes de los 30 años de edad con miocardiopatía, hipogonadismo, diabetes, arritmias e insuficiencia cardíaca. El hierro se acumula mucho más acusadamente que en las hemocromatosis de adultos, distribuyéndose en el hígado predominantemente en los hepatocitos, al igual que en las hemocromatosis tipo 1 y 347. Responde muy bien al tratamiento con flebotomías.
Hay dos subtipos. La tipo 2, subtipo A, u OMIM 602390, se debe a mutaciones en el gen HJV, localizado en el cromosoma 1q2148, que produce la hemojuvelina49, y suelen ser menos graves que las debidas a mutaciones del HAMP. La hemojuvelina se detecta en mutaciones del HJV producen unos valores inadecuadamente bajos de hepcidina, apoyando la hipótesis del papel que desempeña esta última en el metabolismo del hierro41-45. La mutación G320V se da en los dos tercios de las mutaciones detectadas del gen HJV49, otras de las más de 24 mutaciones descritas son la I222N, la L101P, la E302K, la S105L, la N372D, la I281T, la A326T, la C80A...50.
La tipo 2, subtipo B, u OMIM 606464, corresponde a mutaciones en el gen HAMP, localizado en el cromosoma 19q13.151, que codifica la hepcidina, la cual desempeña como hemos citado en varias ocasiones un papel clave en el metabolismo del hierro, y en la actividad antimicrobiana41-46. Entre las mutaciones identificadas en este gen están la 93delG, la G71D, la R59G, la R56X y la C70R52, algunas asociadas a mutaciones de HFE.
Hay que resaltar pues que la hemocromatosis juvenil no es una entidad monógenica invariablemente debida a mutaciones del HJV o del HAMP, sino que puede estar ligada a mutaciones de genes de hemocromatosis hereditarias del adulto53,54.
La hemocromatosis relacionada con el TfR2, o tipo 3, u OMIM 604250, se debe a mutaciones del gen TfR2 localizado en el cromosoma 7q2255, que también está implicado en la captación de hierro por los hepatocitos. Los TfR2 se expresan principalmente en los hepatocitos, aunque también en las criptas duodenales y células eritroides; tienen una menor capacidad (unas 25-30 veces) para acoplar hierro que los TfR17. El HFE y el TfR2 se disponen juntos en las células de las criptas, pero la distribución del TfR2 en las criptas es diferente del TfR1. Es una enfermedad autosómica recesiva, poco frecuente, con un potencial para lesionar órganos similar a la hemocromatosis HFE; suele manifestarse a los 40-50 años de edad, el hierro se deposita en el hígado preferentemente en los hepatocitos y responde satisfactoriamente a las flebotomías. Entre las mutaciones detectadas están la Y250X en 6 pacientes de dos familias italianas55, la M172K y la E60X56, la R455Q, la V2211, la Q690P... Por tanto, un cribado para mutaciones TfR2 puede ser útil en pacientes con sobrecarga férrica que no tienen mutaciones HFE, o que presentan genotipos HFE en heterocigosis.
La hemocromatosis relacionada con la ferroportina o tipo 4, u OMIM 606069, está en relación con mutaciones del gen SLC11A3, localizado en el cromosoma 2q32, que codifica la ferroportina 157-59. Es una enfermedad de carácter autosómico dominante. Suele manifestarse en la cuarta o quinta década de la vida. La ausencia de ferroportina 1, que, como ya hemos mencionado, es una proteína exportadora de hierro13, induce una acumulación de hierro preferentemente en el sistema retículo-endotelial del hígado y del bazo, y menor en los hepatocitos y, posiblemente, de los enterocitos, reduciendo la disponibilidad de hierro para acoplarse a la Tf, y ulteriormente para suministrarlo a la médula ósea, lo que explica los valores bajos o normales de saturación de la Tf; incluso en estadios avanzados de la enfermedad, puede detectarse en ocasiones anemia ferropénica, sobre todo en mujeres posmenárquicas y en personas sometidas a flebotomías. Las manifestaciones fenotípicas son más ligeras que en otros tipos de hemocromatosis, posiblemente porque el daño potencial de órganos es menor cuando el hierro se acumula más en el sistema del retículo endotelial que en las células parenquimatosas. La clave para el diagnóstico no es la determinación del índice de saturación de la Tf sino de la ferritina, la cual puede estar ya elevada en la primera década de la vida, y permanecer alta incluso después de las flebotomías. Las flebotomías frecuentes pueden inducir anemia, por lo que los valores de hemoglobina deben monitorizarse con frecuencia. Se han detectado las mutaciones N144H58, A77D59, Vl62del60, Y64N61, N144T..., algunas en heterocigosis con mutaciones del HFE.
En la hemocromatosis neonatal hay una combinación de sobrecarga hepática masiva de hierro y una insuficiencia hepática perinatal, siendo incierta su índole hereditaria, pues aún se desconoce la localización del gen mutado y la proteína involucrada.

