




El plasmocitoma gástrico es una forma rara de presentación de las gammapatías monoclonales, y puede ser una causa de hemorragia digestiva alta si se encuentra ulcerado. Suele ser localizado, pero puede evolucionar hacia un mieloma múltiple u otras formas de gammapatía monoclonal. El diagnóstico diferencial hay que hacerlo con otras gammapatías monoclonales, basándose en criterios analíticos e histológicos, y con otros tumores gástricos, como el adenocarcinoma, los tumores de la estroma gastrointestinal, los tumores neuroendocrinos y los linfomas. Para ello, nos podemos basar en las diferentes características radiológicas y endoscópicas, aunque, sin duda, el estudio histológico y las técnicas inmunohistoquímicas establecerán el diagnóstico definitivo. En esta observación clínica se describe el caso de un paciente que presentaba una hemorragia digestiva alta y una anemia ferropénica, con diagnóstico final de mieloma múltiple asociado a plasmocitoma gástrico.
Gastric plasmacytoma is an infrequent form of presentation of monoclonal gammopathy that, if ulcerated, can cause upper gastrointestinal bleeding. This entity is usually localized but may progress to multiple myeloma or other forms of monoclonal gammopathy. Differential diagnosis should be performed with other monoclonal gammopathies, based on laboratory and histological criteria, as well as with other gastric tumors such as adenocarcinoma, gastrointestinal stromal tumors, neuroendocrine tumors and lymphomas. Differential diagnosis can be based on radiological and endoscopic characteristics, although definitive diagnosis will undoubtedly be based on histological study and immunohistochemical techniques. We describe the case of a male patient who underwent investigations for upper gastrointestinal bleeding and iron deficiency anemia. The final diagnosis was multiple myeloma associated with gastric plasmacytoma.
El plasmocitoma extramedular (PEM) es un tumor infrecuente formado por células plasmáticas, que crece fuera de la médula ósea, y puede estar asociado o no a un mieloma múltiple (MM). La afectación gastrointestinal representa un 5% de los casos1–6, y las localizaciones más frecuentes son el intestino delgado y el estómago3,4,7. En la mayoría de los casos el PEM es de afectación local, si bien puede progresar excepcionalmente a MM o a otras formas de gammapatía monoclonal5. El PEM gástrico puede ser asintomático o bien producir anorexia, pérdida de peso, dolor abdominal, vómitos o, más raramente, hemorragia digestiva alta por necrosis o ulceración del tumor4,6,7.
OBSERVACIÓN CLÍNICAVarón de 64 años de edad, con antecedentes de enfisema bulloso y adenocarcinoma de recto (T3N0M0), tratado con radioquimioterapia neoadyuvante y cirugía 3 años antes. Su madre había fallecido por un adenocarcinoma de colon y su padre por un adenocarcinoma de próstata. Dos meses antes del ingreso en nuestro servicio fue diagnosticado de una anemia normocítica y normocrómica, atribuida a un origen multifactorial. En el momento del ingreso en nuestro servicio, el paciente refería clínica dispéptica, síndrome constitucional y deposiciones melénicas de unos 20 días de evolución, así como un dolor intenso en el hipocondrio izquierdo irradiado a la fosa renal, que le obligaba a tomar derivados mórficos para su control. En la exploración física se palpaba una adenopatía supraclavicular izquierda y otra laterocervical derecha; el resto de la exploración fue irrelevante.
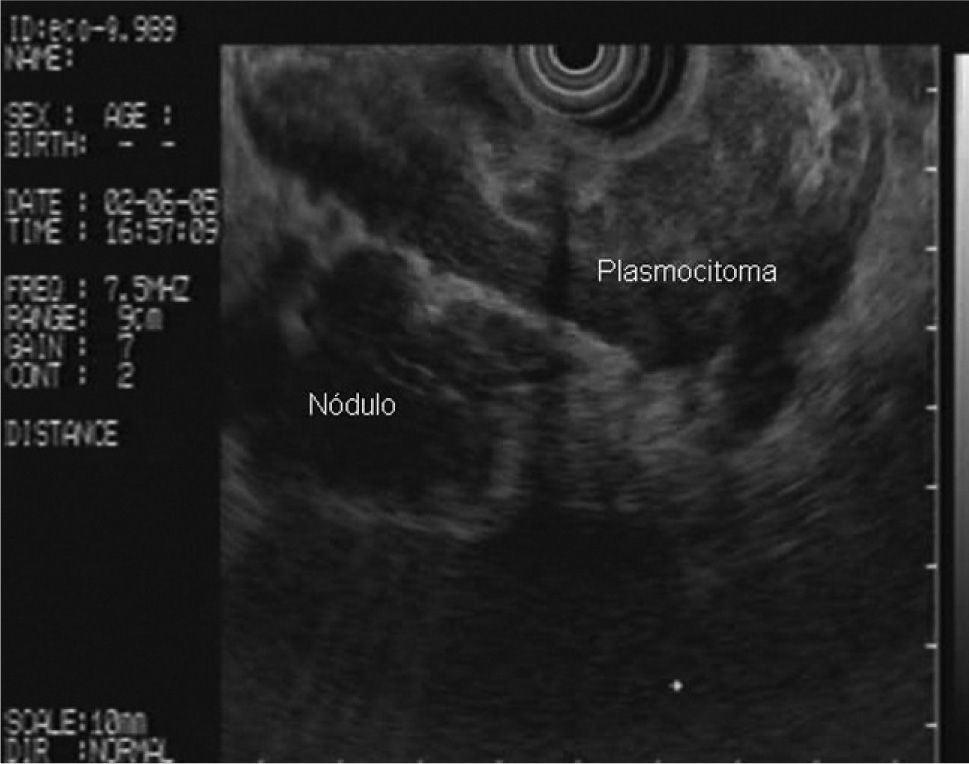
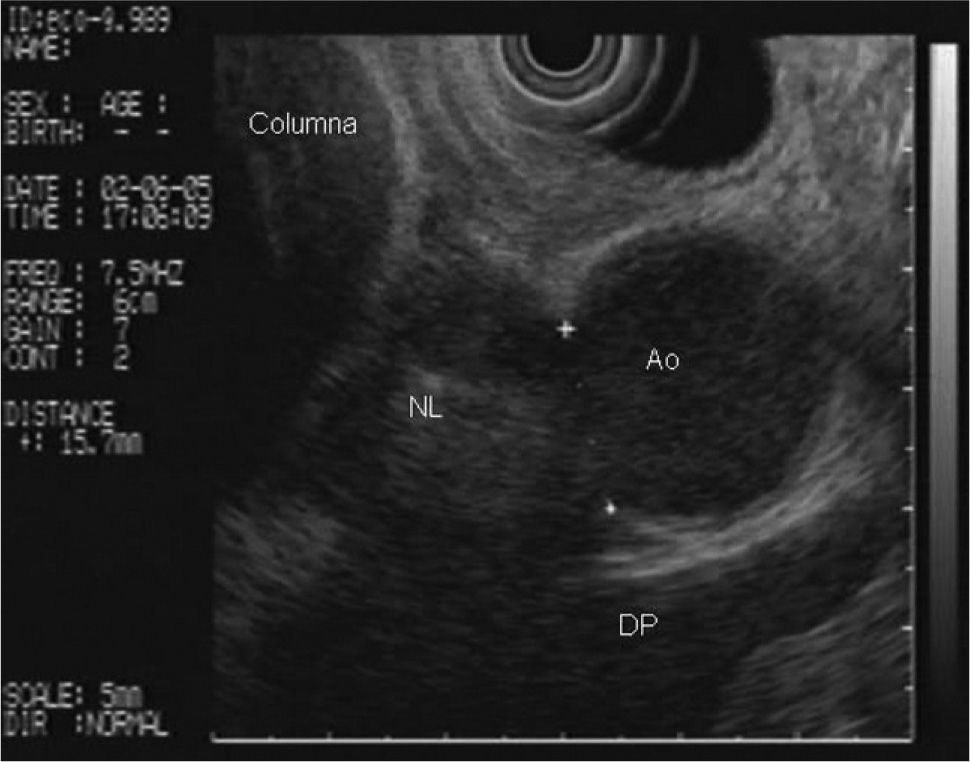
En la analítica de sangre destacaban los siguientes parámetros: hemoglobina de 4,8 g/dl (rango, 13-18), hematocrito del 14,5% (rango, 38- 50), volumen corpuscular medio de 97,7 μ3 (rango, 80,2-99,4), urea de 86,8 mg/dl (rango, 10-50) y creatinina de 2,1 mg/dl (rango, 0,6-1,2); el resto de los parámetros bioquímicos estaban dentro de la normalidad. Se realizó una gastroscopia de urgencia, en la cual se puso de manifiesto una neoformación vegetante subcardial, ulcerada, con restos hemáticos en su superficie y sin sangrado activo. Se practicó una ecoendoscopia, en la que se observó el tumor como un engrosamiento heterogéneo (de hasta 2,5 cm) de la pared en el cuerpo gástrico proximal, con afectación de todas las capas, en mayor medida de la muscular propia, y sobrepasando la serosa, con afectación de la grasa perigástrica (fig. 1). Además, se identificaron múltiples adenopatías de gran tamaño (2,5-3 cm), hipoecoicas, bien delimitadas, en el hilio esplénico, el área pancreática, el área de tronco celíaco, el hilio hepático y el mediastino (fig. 2
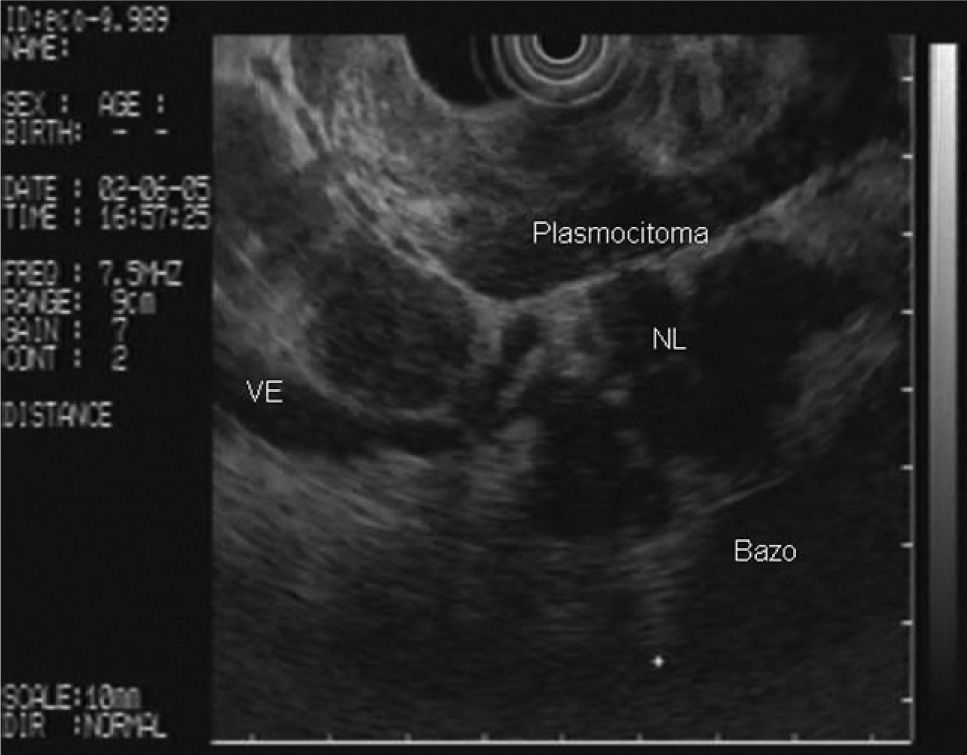
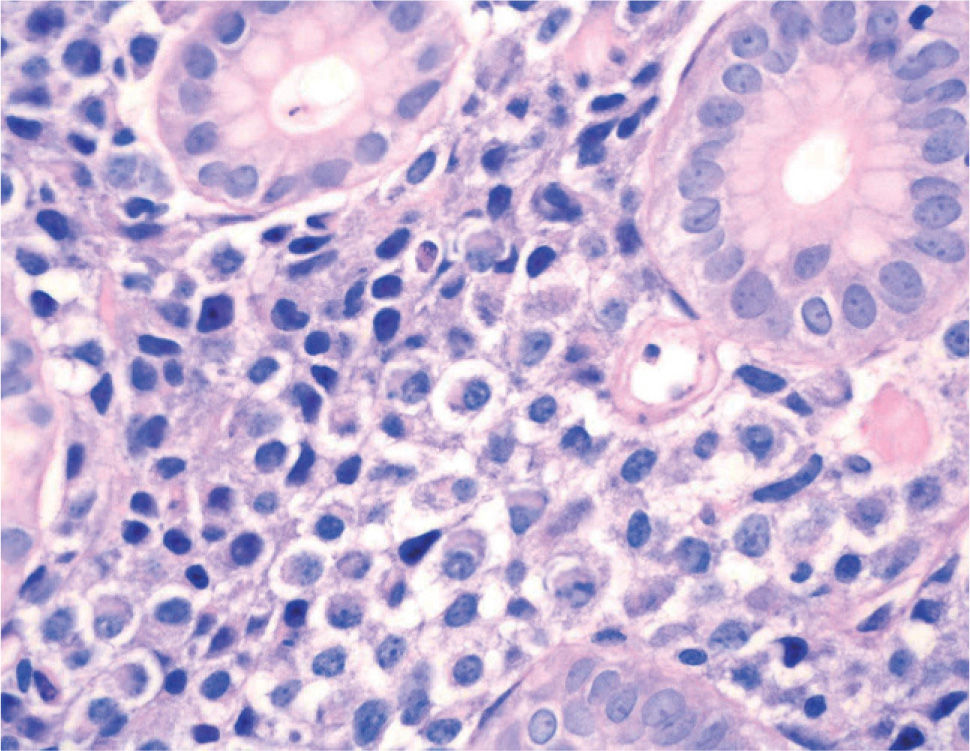
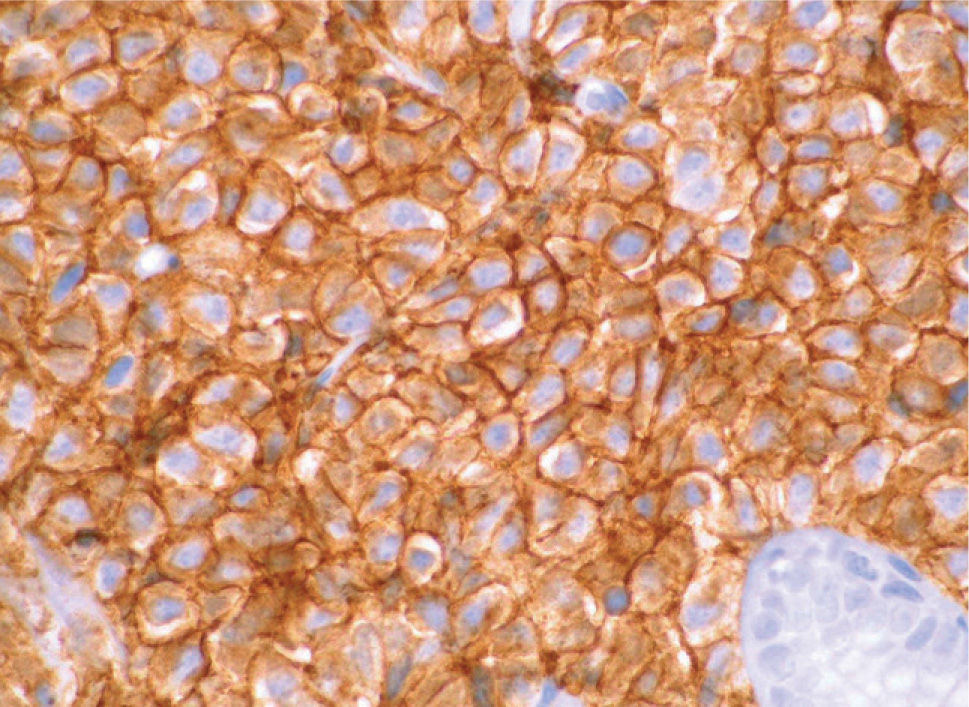
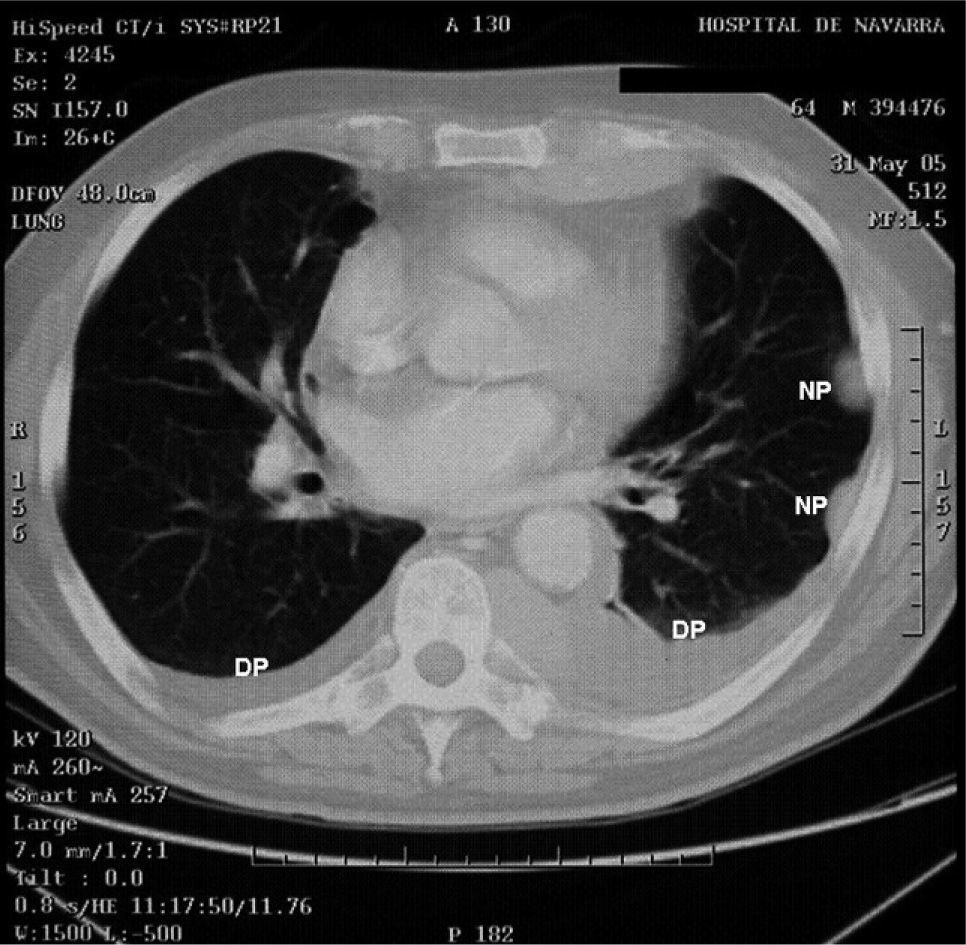
). El estudio anatomopatológico de las biopsias gástricas demostró una infiltración masiva de la lámina propia por células redondas, con un citoplasma finamente granular y núcleos redondeados u ovoides, que expandía el espacio entre las estructuras glandulares y presentaba una moderada variabilidad en el tamaño nuclear y un alto recambio celular (fig. 4). Las técnicas inmunohistoquímicas determinaron negatividad para CD20, CD45, CD5 y CD23 y positividad para CD56, CD38, cadena lambda, Bcl-2, ciclina D1, EMA y p53, todo ello compatible con un plasmocitoma gástrico (fig. 5). Asimismo, se biopsió la adenopatía supraclavicular izquierda, con un estudio anatomopatológico superponible al de las biopsias gástricas. En el estudio de extensión mediante tomografía computarizada (TC) se observaron numerosas lesiones nodulares pleurales, principalmente en el hemitórax izquierdo, con diámetro de hasta 6 cm, acompañadas de un derrame pleural, que se llegaban a introducir por los espacios intercostales, así como adenopatías sobre la cola del páncreas y adyacentes al hilio esplénico (fig. 6). En la gammagrafía ósea se apreciaban focos de hiperfijación en el húmero derecho, las costillas izquierdas 3 y 7, la región frontal izquierda y el tobillo derecho, compatibles con metástasis. Se realizó un medulograma, que mostró una infiltración de la médula ósea por un 31% de células plasmáticas de maduración intermedia. Se completó el estudio analítico constatando en suero una banda monoclonal de cadena ligera λ, sin cadena pesada asociada, con déficit de todos los subtipos de inmunoglobulinas, y proteinuria de Bence- Jones positiva en orina de 24 h. Ante todos estos hallazgos, el paciente fue diagnosticado de MM en estadio IIIA (cadenas ligeras λ, Bence-Jones positivo) y hemorragia digestiva alta secundaria a un plasmocitoma gástrico ulcerado. Durante el ingreso se trasfundieron 10 concentrados de hematíes, y el paciente comenzó tratamiento con pamidronato i.v. y dexametasona durante 4 días. Tras comprobar que no había signos de resangrado, se inició un tratamiento alternante con una pauta combinada de vincristina, carmustina, melfalan, ciclofosfamida y prednisona, y otra de vincristina, carmustina, merfalan, dexametasona, junto con somatostatina i.v. en infusión continua para profilaxis de la hemorragia digestiva alta. No se constató una respuesta al tratamiento de las lesiones. A los 3 meses se cambió el tratamiento a bortezomib-dexametasona, con lo que el paciente presentó inicialmente una respuesta parcial, con posterior progresión de la enfermedad, por lo que se cambió el tratamiento a un protocolo poliquimioterápico con ifosfamida, carboplatino y etopósido. Ocho meses después el paciente presentó una encefalopatía multifactorial, un síndrome febril sin foco y disnea, y falleció a los pocos días.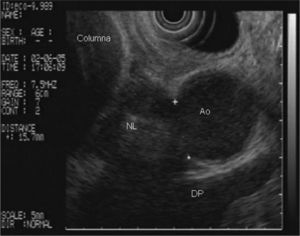
.")
, así como el bazo y un conglomerado adenopático en hilio esplénico (NL).")
.")
.")
 izquierdos, junto con derrame pleural (DP) bilateral.")
El PEM es una entidad poco frecuente de tumores de células plasmáticas, que supone menos del 5% de todos ellos2,6. Dentro de los PEM, un 5% es de localización gástrica exclusiva3,4,8. Sin embargo un 15% de los PEM puede evolucionar a una forma generalizada, como el MM1, que tiene una incidencia anual de 5 casos por 100.000 habitantes9. El MM generalmente afecta sólo a la médula ósea, aunque un 20% de los casos puede presentar una lesión en otras localizaciones, como las vías respiratorias altas o el tracto gastrointestinal4. Por tanto, en nuestro paciente cabían las 2 posibilidades: que se tratara de un plasmocitoma gástrico evolucionado a MM, o un MM con afectación extramedular. Para llegar al diagnóstico de MM se deben cumplir determinados criterios, que consisten en la presencia de proteína M en sangre u orina, células plasmáticas monoclonales en la médula ósea (> 10%) y, eventualmente, lesiones osteolíticas o masas extramedulares1, presentes en nuestro paciente.
Inicialmente, ante el hallazgo de la tumoración gástrica, se planteó además el diagnóstico diferencial con otros tumores gástricos: adenocarcinoma, tumores del estroma gastrointestinal (GIST), tumores neuroendocrinos y linfomas7,10,11. Endoscópicamente, el PEM puede presentarse en diferentes formas –masa aislada, tumoración ulcerada, como es el caso de nuestro paciente, o varias ulceraciones mucosas12– y esta misma morfología puede aparecer en los otros tipos de tumores. En la imagen de la TC, el plasmocitoma también puede ser parecido a todos ellos, ya que todos se caracterizan por un engrosamiento homogéneo de la pared gástrica7, aunque se han descrito ciertas diferencias que nos permitan distinguir entre un tipo de tumor u otro. Así, en los linfomas, las adenopatías son mucho más voluminosas que en los plasmocitomas. Los GIST y los tumores neuroendocrinos, en cambio, no suelen mostrar adenopatías, y frecuentemente presentan necrosis intratumoral3,7,10,13. El reto ante un plasmocitoma gástrico consiste en diferenciarlo de un linfoma. Hay un 30% de linfomas del tejido linfoide asociado a las mucosas (MALT) con diferenciación plasmocítica, indistinguible histológicamente de un plasmocitoma, sobre todo un tipo infrecuente de linfoma MALT, denominado enfermedad inmunoproliferativa del intestino delgado14. El estudio anatomopatológico y las técnicas inmunohistoquímicas establecerán el diagnóstico diferencial entre ambas7. Otra entidad que hay que tener en cuenta a la hora del diagnóstico diferencial son los linfomas plasmoblásticos con diferenciación plasmocelular. Son linfomas morfológica e inmunohistoquímicamente indistinguibles de la variante plasmoblástica de los PEM. En la práctica, la distinción entre ambos se basa en la correlación clínica. La presencia de una banda monoclonal en suero y/o lesiones líticas óseas en las pruebas de imagen apoyarían el diagnóstico de MM. Sin embargo, la presencia de infección por el virus de Epstein-Barr o el virus de la inmunodeficiencia humana apoyarían el diagnóstico de linfoma plasmoblástico15,16.
Al igual que otros tumores gástricos17, en la literatura médica se han descrito casos de plasmocitomas gástricos asociados a la presencia de Helicobacter pylori, si bien esta relación no está clara2. En algunos de estos casos se describe que la erradicación de dicha bacteria lleva a la regresión completa del tumor, tanto endoscópica como anatomopatológica2, aunque en algún otro caso, la regresión es sólo endoscópica3,8. Sin embargo, dada la dificultad para el diagnóstico histológico de plasmocitoma, se ha planteado que los casos descritos de plasmocitomas que remiten tras tratamiento erradicador de H. pylori se tratan realmente de linfomas MALT con gran diferenciación plasmocítica14.
Mediante ultrasonografía endoscópica no hay criterios establecidos para poder diferenciar el plasmocitoma del linfoma gástrico. Dada la baja frecuencia del primero y las características ecográficas similares de ambos, parece poco probable que con la tecnología actual se puedan llegar a diferenciar mediante esta técnica.
Actualmente se recomienda un tratamiento basado en la radioterapia para el PEM localizado, que puede conseguir un control local o incluso la curación de este tipo de tumores. Sin embargo, en los casos muy avanzados se recomienda un tratamiento quimioterápico similar al del MM1,4–6. Al presentar nuestro paciente un estadio avanzado de la enfermedad en el momento del diagnóstico, se optó por la segunda opción de tratamiento, aunque no se obtuvo una respuesta favorable.

