




La porfiria aguda intermitente es un trastorno hereditario autosómico dominante producido por un déficit en la actividad enzimática de la porfobilinógeno deaminasa, la tercera enzima en la síntesis del hemo. Se trata de una enfermedad rara, aunque los portadores heterocigotos asintomáticos tienen una prevalencia mayor, difícil de establecer dada la ausencia de clínica. Aunque la porfiria aguda intermitente es una enfermedad multisistémica, su presentación más frecuente es el dolor abdominal y los síntomas neurológicos o psiquiátricos, a veces debidos a algunos factores precipitantes, como la baja ingesta energética, el tabaco, el alcohol, algunos fármacos y el estrés. El diagnóstico se puede realizar midiendo el nivel de porfobilinógeno urinario, con posterior análisis de la actividad de la enzima defectuosa y estudio del ADN. El tratamiento se basa en la prevención de los ataques de porfiria, evitando los factores desencadenantes, y en la administración precoz de glucosa i.v. o tratamiento con hematina. Presentamos el caso de una paciente diagnosticada de porfiria aguda intermitente a partir del estudio de una elevación crónica leve de la GPT.
Acute intermittent porphyria is an autosomal dominant in-herited disorder resulting from a deficiency of porphobilinogen deaminase activity, the third enzyme in the heme biosynthesis pathway. This disease is uncommon, although the prevalence is higher in asymptomatic heterozygotic carriers; however, this prevalence is difficult to establish be-cause of the absence of symptoms. Although acute intermittent porphyria is a multisystemic disease, its most common form of presentation is abdominal pain and neurological or mental symptoms, which can sometimes be due to precipitating factors such as reduced energy intake, smoking, alcohol, some drugs, and stress. Diagnosis can be made by testing urinary porphobilinogen levels, with subsequent measurement of enzyme activity and DNA testing. Treatment is based on prevention of porphyria attacks by avoiding precipi-tating factors and early administration of intravenous glucose or hemin therapy. We present the case of a patient diagnosed with acute intermittent porphyria based on study of chronic mild alanine aminotransferase (ALT) elevation.
Las porfirias son trastornos hereditarios o adquiridos de la síntesis del hemo1. Ésta se realiza en su mayor parte en las células eritroides y el resto en el hígado. La aminolevulinato sintetasa es la primera enzima en la síntesis del hemo y su actividad está regulada por un sistema de retroalimentación negativa. Las porfirias hereditarias clásicamente se han dividido en hepáticas y eritropoyéticas, según el lugar donde se acumulan las porfirinas y sus precursores; la clínica neurológica es la característica principal de las hepáticas, y la afectación cutánea la de las eritropoyéticas. Otra clasificación las divide en agudas o crónicas, en función de sus signos y síntomas, aunque en la actualidad tienden a clasificarse por la enzima defectuosa. Presentamos el caso de una paciente con porfiria aguda intermitente diagnosticada a partir del estudio de una hipertransaminasemia prolongada.
OBSERVACIÓN CLÍNICAMujer de 50 años de edad, que consulta por presentar desde 1998 una discreta elevación de la GPT (valores siempre inferiores a 1,5 veces el valor normal, entre 32 y 43 U/l en 10 determinaciones), con GOT y GGT normales. Entre sus antecedentes familiares destacan los siguientes: padre con cáncer de colon y abuela paterna fallecida por hepatopatía no filiada a los 35 años. Refiere antecedentes personales de hepatitis A en 1988, cistitis de repetición desde el año 2000, dos partos, síncope en el primero tras recibir anestesia, menopausia en 2005 y un cuadro de hipotensión e hipoglucemia tras administrar anestésico tópico para una ci- rugía menor. Desde 1993, tras el fallecimiento de su marido, presenta molestias abdominales difusas, ocasionalmente intensas, consistentes en dolor en el flanco izquierdo y estreñimiento, por lo que se le realizó un enema opaco en 1995, una colonoscopia izquierda en 1996 y un nuevo enema opaco en 1999, sin lesiones. En 2005 se le practicó una colonoscopia total, dados sus antecedentes familiares, en la que sólo se observaron hemorroides internas. El mismo año consultó por un cuadro de fiebre, mialgias, astenia y orina oscura, que remitió en 2-3 días.
En la exploración física se observó que la paciente estaba normosómica, con una presión arterial de 110/70 mmHg; el tórax, el abdomen y los miembros no presentaban anomalías.
En la analítica practicada presentó unas cifras de colesterol total de 220 mg/dl y GPT de 34 U/l. El hemograma y las cifras de glucosa, creatinina, triglicéridos, acido úrico, TSH, T4, GOT, GGT, bilirrubina, hierro, ferritina, transferrina, alfafetoproteína, IgG, IgM, IgA, ceruloplasmina, cupremia, cupruria y alfa-1 antitripsina eran normales. La serología del VHB y el VHC, así como los ANA, AMA, AML y anti-LKM eran negativos. En el estudio de las porfirinas en orina de 24 h, los valores de uroporfirinas eran de 269 μg/24 h, y en una segunda determinación de 276 μg/24 h (valor normal < 50 μg/24 h), las coproporfirinas de 159,86 μg/24 h y en una segunda determinación de 371,49 μg/24 h (valor normal < 150 μg/24 h), el ácido delta aminolevulínico (ALA) de 10,5 mg/24 h (valor normal < 7 mg/24 h) y el porfobilinógeno (PBG) de 16,88 mg/24 h (valor normal < 2 mg/24 h). En el estudio enzimático los valores de PBG deaminasa eritrocitaria eran de 13,3 nmol/h/ml eritrocitos (valor normal, 25-44 nmol/h/ml eritrocitos), y presentaba un déficit diagnóstico de porfiria aguda intermitente (PAI). El estudio genético directo de la región codificante del gen HMBS revelaba que la paciente era portadora heterocigota de una C inserción en el exón 14, probable causante de PAI. La ecografía abdominal era normal.
Se estudió a sus 2 hijos: uno de ellos presentaba una PBG deaminasa eritrocitaria de 12,56 nmol/h/ml eritrocitos y el otro de 15,24 nmol/h/ml eritrocitos. En el estudio genético se observó que ambos presentaban la misma alteración que la madre.
La paciente fue derivada a una unidad especializada en porfirias para su seguimiento. No se practicó biopsia hepática ante la elevación mínima de la GPT, y se recomendó mantener una actitud expectante y asesoramiento de la paciente y de sus hijos sobre la enfermedad, así como evitar los posibles desencadenantes de un ataque de porfiria.
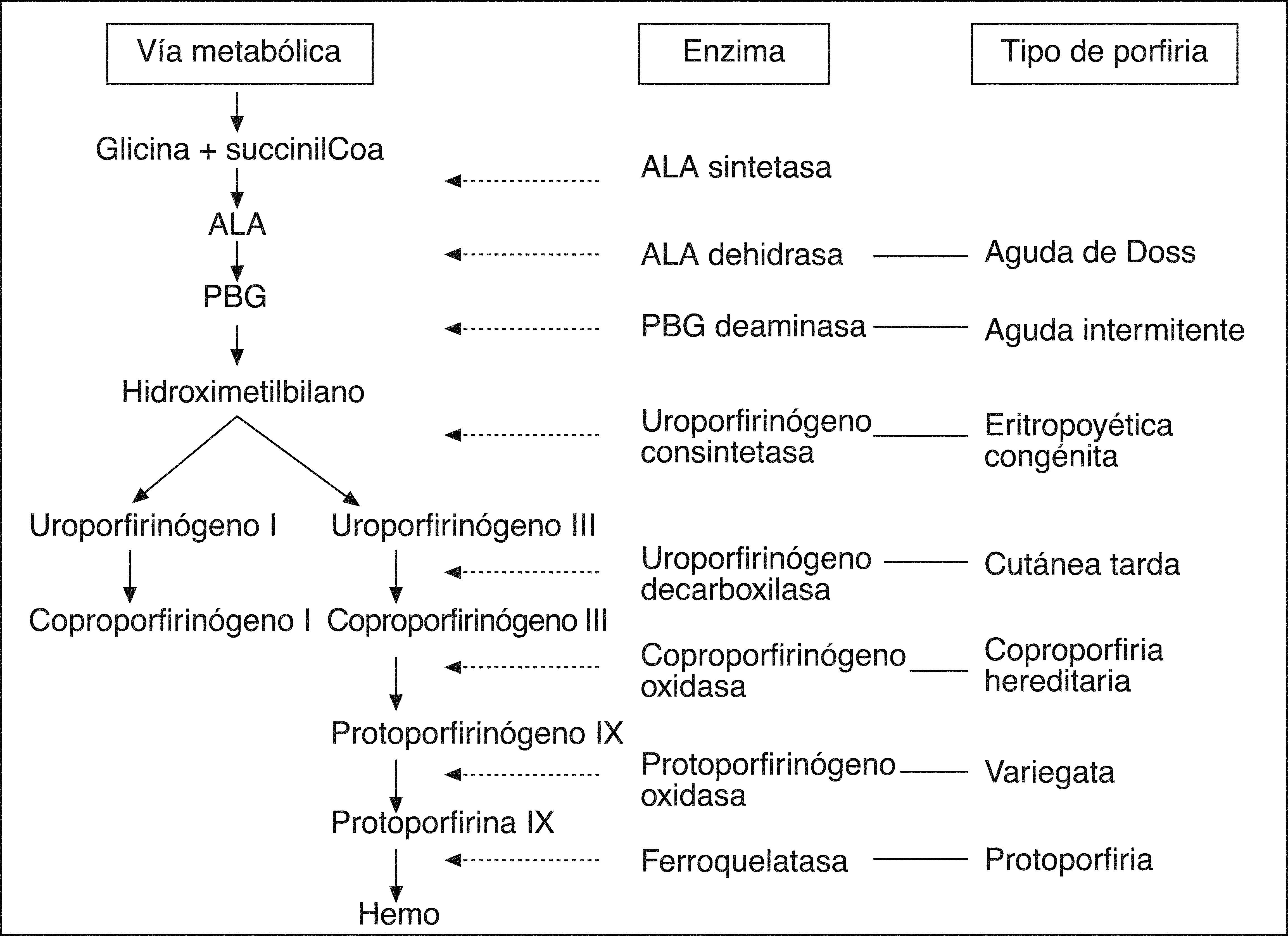
DISCUSIÓNLa PAI está considerada como la más grave de las porfirias hepáticas hereditarias. Es frecuente en los países nórdicos, como Suecia, pero es rara en el resto de Europa (1-2/100.000)2. En España hay pocos casos publicados. Es difícil conocer su prevalencia real, puesto que la mayoría de los pacientes son heterocigotos y están asintomáticos. Su transmisión es autosómica dominante3. La clínica surge tras una reducción a la mitad de la actividad de la enzima PBG deaminasa, también denominada uroporfirinógeno sintetasa o hidroximetilbilano sintetasa4. Hay 2 isoenzimas de la PBG deaminasa, una eritroide específica y otra no eritroide. Dependiendo de la localización de la mutación, pueden faltar sólo una o las 2 isoenzimas, por lo que su valor puede ser normal en los eritrocitos en algunos pacientes afectados. Cuando se produce un déficit, éste es parcial al 50%, y es suficiente para la homeostasis del hemo en condiciones normales5 (fig. 1). Las crisis de porfiria se desencadenan cuando actúan agentes externos precipitantes que aumentan la demanda de la síntesis hepática de hemo e inducen la actividad de la ALA sintetasa, como el estrés, el tabaco, las dietas hipocalóricas, los factores hormonales, la ingesta de fármacos (barbitúricos, anticonvulsivantes, rifampicina…), las enfermedades asociadas y la ingesta de alcohol6,7. Todo ello, unido al déficit enzimático de la PBG deaminasa, produce una acumulación y un depósito tisular de los precursores ALA y PBG (fig. 1). Por tanto, hay déficit de hemo y acumulación de precursores que son tóxicos directa o indirectamente. En la PAI no hay manifestaciones de fotosensibilidad porque los precursores acumulados son inactivos al interaccionar con la luz solar. Sí son neurológicamente tóxicos, tanto en el sistema nervioso central como en el sistema nervioso autónomo y periférico. Muchos pacientes pueden no tener los precursores elevados y, aunque el 90% de ellos están asintomáticos, al exponerse a los factores precipitantes pueden tener una crisis.

Los síntomas neuroviscerales suelen aparecer después de la pubertad y son más frecuentes en las mujeres. La disfunción del sistema nervioso autónomo puede ocasionar dolor abdominal, náuseas, vómitos, diarrea o estreñimiento, e incluso distensión e íleo. El dolor abdominal intenso obliga a realizar un diagnóstico diferencial con la apendicitis aguda, aunque no suele asociar signos de irritación peritoneal. El dolor abdominal en el contexto de un cuadro estresante, muchas veces el desencadenante, conduce con frecuencia al diagnóstico erróneo de histeria. También pueden asociarse molestias o retención urinaria, sudoración, hipertensión, cansancio y temblores por hiperactividad simpática, así como hiponatremia por un síndrome de secreción inadecuada de ADH. La afectación del sistema nervioso periférico se manifiesta como una polineuropatía proximal de predominio motor y, con el tiempo, los pacientes pueden presentar una afectación distal sensitivo-motora (axonal y desmielinizante proximal y distal en el electromiograma)8. A veces presentan debilidad muscular, espasmos, calambres, alteración de reflejos y progresión a parálisis respiratoria con afectación bulbar, coma y muerte. El ataque puede durar horas, con una recuperación más rápida del dolor abdominal que de la paresia. En ocasiones, las porfirias agudas presentan únicamente clínica psiquiátrica, como ansiedad, depresión e, incluso, agitación, delirio, alucinaciones y convulsiones9.
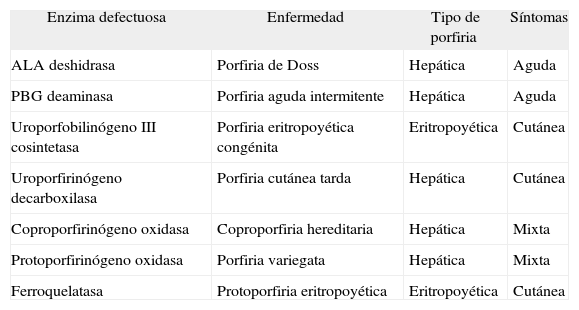
El diagnóstico debe sospecharse en los pacientes con un dolor abdominal recurrente, en quienes no se encuentra la causa tras realizar las exploraciones complementarias habituales10. A veces se somete al paciente a técnicas de imagen e incluso procedimientos quirúrgicos innecesarios, cuando una simple determinación de los valores de PBG en orina podría establecer el diagnóstico, aunque resulta inespecífico del tipo de porfiria. En los ataques aumentan la ALA y el PBG en orina y, en los casos graves, ésta adquiere un color rojo vinoso, debido al alto contenido en porfobilina, producto de la degradación del PBG. Se dispone de tests rápidos para detectar los valores de PBG urinario. En las demás situaciones se utiliza la medición de ALA y PBG en orina de 24 h10,11. Hay que realizar el diagnóstico diferencial con la porfiria variegata (PV) y la coproporfiria hereditaria (CH), que pueden presentar ataques neurológicos y elevación de PBG y ALA en orina1 (tabla I). La PAI no cursa con fotosensibilidad, a diferencia de la PV y la CH, en las que puede haber síntomas cutáneos, como consecuencia de la acumulación de protoporfirina y coproporfirina, respectivamente (fig. 1). La excreción de coproporfirina está elevada en orina y heces en la CH, pero es normal o escasamente elevada en la PV y la PAI, y la excreción de protoporfirina en heces está elevada en la PV, pero es normal en la CH y la PAI. La actividad de la PGB deaminasa es normal en la CH y la PV.
Clasificación de las porfirias
| Enzima defectuosa | Enfermedad | Tipo de porfiria | Síntomas |
| ALA deshidrasa | Porfiria de Doss | Hepática | Aguda |
| PBG deaminasa | Porfiria aguda intermitente | Hepática | Aguda |
| Uroporfobilinógeno III cosintetasa | Porfiria eritropoyética congénita | Eritropoyética | Cutánea |
| Uroporfirinógeno decarboxilasa | Porfiria cutánea tarda | Hepática | Cutánea |
| Coproporfirinógeno oxidasa | Coproporfiria hereditaria | Hepática | Mixta |
| Protoporfirinógeno oxidasa | Porfiria variegata | Hepática | Mixta |
| Ferroquelatasa | Protoporfiria eritropoyética | Eritropoyética | Cutánea |
ALA: ácido deltaaminolevulínico; PBG: porfobilinógeno.
El tratamiento se basa fundamentalmente en la prevención de los ataques, instruyendo a los pacientes o portadores para evitar los factores desencadenantes. En el ataque agudo se deben tratar las posibles infecciones, así como la hipertensión, el dolor y la hiponatremia. En los casos graves deben administrarse hidratos de carbono intravenosos, al menos 400 g/día. A veces es necesaria la nutrición parenteral. El dolor se puede tratar con narcóticos, y las benzodiacepinas en dosis bajas son seguras. El uso de preparados intravenosos de hemo (hematina o arginato de hemo) disminuye la actividad de la ALA sintetasa. Se administra en dosis de 3-5 mg/kg durante al menos 4 días10, y es más eficaz si su uso es precoz. Se han comunicado ataques agudos tratados con cimetidina en dosis de 300 mg cada 6 h, con resolución a las 72 h y, en algunos casos se ha observado un período de remisión más prolongado con dosis de 400-800 mg/día en tratamiento preventivo, aunque su indicación no está plenamente establecida. Puede considerarse su uso en el ataque agudo si no se dispone de hematina. Se cree que actúa disminuyendo el consumo de hemo e inhibiendo la ALA sintetasa a través de un feed-back negativo12.
La paciente del caso descrito no presentó alteraciones del comportamiento ni clínica psiquiátrica, salvo algún síntoma de ansiedad que creyó debido a la menopausia. El dolor abdominal que manifestó no es característico de un ataque agudo, pues se trata de un dolor difuso y mal delimitado, con escasas fluctuaciones a lo largo de los años. Sin embargo, no podemos excluir este diagnóstico en alguna ocasión que presentó un dolor más intenso y en el episodio de fiebre, mialgias y orinas oscuras que la paciente había relatado, aunque también pudo tratarse de un cuadro febril autolimitado con orina concentrada. Con respecto a los síntomas relacionados con las anestesias, es dudoso que se debieran a la porfiria, ya que no hubo manifestaciones posteriores y la resolución fue rápida y espontánea. La paciente refiere el antecedente del fallecimiento de una de sus abuelas por un problema hepático, pero no podemos atribuirlo a esta enfermedad, porque no conoce más datos. Es muy difícil distinguir en nuestra paciente si la elevación de la GPT es debida a la PAI o a otra enfermedad hepática no relacionada. De hecho, cuando se observó inicialmente una elevación de las porfirinas en orina se sospechó una porfiria hepatocutánea tarda por ser la más frecuente y manifestarse después de la cuarta década de la vida, aun en ausencia de síntomas cutáneos. No obstante, se ha descrito algún caso de PAI con notable elevación de las transaminasas en un episodio de dolor abdominal que requirió hospitalización13, y está documentado el riesgo de carcinoma hepatocelular en los pacientes con porfiria14–16. Además, en algunas series de pacientes con PAI asintomáticos se detectan, hasta en un 13% de los casos, alteraciones en la GPT17. Sin embargo, no se han comunicado casos de PAI o de portadores de esta enfermedad diagnosticados a partir de una elevación tan leve y persistente de la GPT. El discreto aumento de la GPT que presentó la paciente no se debió a factores habituales (ingesta de alcohol, fármacos, obesidad o dislipemia) y se descartaron otras causas de hepatopatía, como virus, alteraciones del metabolismo del hierro y cobre, déficit de alfa-1 antitripsina y trastornos autoinmunes. Probablemente, una biopsia hepática habría ayudado a filiar definitivamente la hepatopatía, pero se decidió mantener una actitud expectante al ser una prueba no exenta de riesgos, presentar una elevación tan discreta de la GPT y optar la paciente por un seguimiento menos agresivo. Por tanto, no se puede descartar que la elevación crónica de la GPT de la paciente se debiera a la PAI. Si bien no se contempla de manera habitual18,19, debe considerarse este diagnóstico diferencial en el estudio de la hipertransaminasemia mantenida, aunque ésta sea leve, y no haya otra causa que lo justifique.

