




IgG4-related disease is a fibrous-inflammatory process related to immunomodulation. The most commonly affected organs are: the pancreas, bile duct, major salivary glands, lacrimal glands, retroperitoneum and lymphatic ducts.
In recent decades, this disease has been recognised as a systemic disorder that includes many single organ disorders, previously unrelated and known as independent entities.
The common characteristics shared by the different entities that make up the IgG4-related disease are: raised serum IgG4 levels, alterations in the imaging tests with neoplastic-like swelling of the affected organs, specific histopathological characteristics and in immunostaining, as well as good response to treatment with glucocorticoids.
In this work, we will review this pathology with a special emphasis on the characteristics of autoimmune pancreatitis, sclerosing cholangitis related to IgG4 and the involvement of the retroperitoneum, mesenterium and the digestive tract.
La enfermedad relacionada con la IgG4 es un proceso fibroinflamatorio relacionado con la inmumomediación. Los órganos que se ven afectados por esta enfermedad más frecuentemente son: el páncreas, la vía biliar, las glándulas salivares mayores, lacrimales, retroperitoneo y linfáticos.
En las últimas décadas esta enfermedad ha sido reconocida como un trastorno sistémico que engloba a muchas afecciones individuales de órganos, antes no relacionadas y conocidas como entidades independientes.
Las características comunes compartidas por las distintas entidades que componen la ER-IgG4 son: cifras elevadas de IgG4 sérica, alteraciones en las pruebas de imagen con tumefacción de aspecto neoplásico de los órganos afectados, características histopatológicas propias y en inmunotinción, buena respuesta al tratamiento con glucocorticoides.
En este trabajo realizaremos una revisión de esta patología con especial énfasis en las características de la pancreatitis autoinmune, colangitis esclerosante relacionada con IgG4, el compromiso del retroperitoneo y mesenterio, y del tubo digestivo.
Immunoglobulin G4-related disease (IgG4-RD) is a fibrous-inflammatory process related to immumomodulation. It can affect multiple organs, causing in them swollen, destructive lesions and organ failure.1,2 The most commonly affected are the pancreas, bile duct, major salivary glands, lacrimal glands, retroperitoneum and lymphatic ducts.
In recent decades, this disease has been recognised as a unified systemic disorder that includes many single organ disorders, previously unrelated and known as different entities.
It is, therefore, a recent disease, of unknown cause, which comprises a wide spectrum of alterations with common pathological, serological and clinical characteristics. It is an underdiagnosed disease mainly because it is poorly understood, being frequently confused with cancer, infection or other autoimmune diseases such as Sjögren's or Wegener's syndrome.
The common characteristics shared by the different entities that make up IgG4-RD are:
- –
Swelling of the neoplastic aspect of the affected organs.
- –
Lymphoplasmocytic infiltrate rich in IgG4-positive plasma cells.
- –
Variable degree of fibrosis with a storiform pattern.
- –
In addition, high levels of serum IgG4 are found in 60–70 % of patients.
- –
The vast majority of patients respond to glucocorticoids, especially in the early stages of the disease.3
In 2001, the association of autoimmune pancreatitis (AIP) with high plasma IgG4 levels was described. This finding thenceforth became a criterion for the diagnosis of type 1 AIP and, over time, for IgG4-RD.1
In 2003 it was observed that patients with type 1 AIP had fibrous-inflammatory lesions rich in both synchronous and metachronous IgG4 cells in other organs. This fact expanded the concept from a local disease to a multiorgan or systemic disease.
Currently, this condition has been described in virtually any organ: pancreas, biliary tree, salivary glands, periorbital tissues, pituitary gland, kidneys, lungs, lymph nodes, meninges, breast, prostate, thyroid, pericardium and skin. All of them show similar histological characteristics. Therefore, the diseases that are part of the IgG4-RD spectrum include: AIP, sclerosing cholangitis, retroperitoneal fibrosis, inflammatory aortic aneurysm and sclerosing mesenteritis.4
The nomenclature of this entity has evolved from IgG4-related multiorgan proliferative syndrome, IgG4-related sclerosing disease, IgG4-related systemic disease until in October 2011 IgG4-related disease was chosen by a group of Japanese researchers. This term, chosen at the 2011 Boston Symposium by experts from different specialties, refers to the ubiquity of IgG4 in the affected organs and its frequently high plasma levels.5
EpidemiologyThe available epidemiological data are biased because they are mainly based on Asian publications. The study of the disease is difficult due to the small quantity of studies and data collected, as there were no publications on this disease until 2003. Japanese studies show a prevalence of about 100 cases per 10,000 people. However, it is known to be underdiagnosed.6
IgG4-RD has a higher prevalence in male patients (60.8–83 %) and in those over 50 years of age.7–12 A male/female ratio of 2.8:1 has been described. However, this data comes from cohorts of patients with autoimmune pancreatitis and Asian populations—as we have already noted—so it cannot entirely be extrapolated to other populations.7
This ratio of men to women in type 1 AIP contrasts with the ratio in classic autoimmune diseases, where the predilection to these diseases for women is 9:1.
No familial cases of IgG4-RD are known at this time. There are also not enough studies that include data such as patients' ethnicity to draw conclusions about genetic susceptibility.3 And no relation to tobacco, alcohol abuse or eating habits has been documented.13
Clinical manifestations and diagnosisAs it is a disease that can affect multiple organs, its clinical manifestations will depend on which organs are affected, and the initial presentation may be nonspecific or indicate other much more common diseases.14
The key to diagnostic suspicion is multiorgan involvement, a previous clinical history of AIP or both. The most accurate evaluation of IgG4-RD is based on a thorough medical history, physical examination, selected laboratory tests, typical histopathology findings and appropriate radiological studies.1,15
The imaging tests used to diagnose this entity are computed tomography (CT) and magnetic resonance imaging (MRI). Positron emission tomography-CT (PET-CT) is also a useful technique in diagnosis. An increase in fluorodeoxyglucose uptake has been demonstrated during the active phase of the disease, with remission after the corticosteroid treatment. It is useful, therefore, for initial staging, to locate the target organ for biopsy and to evaluate its response to treatment.
The measurement of SUVs (standard uptake values) between the pancreas and the liver could be useful for differentiating AIP and cancer according to recent studies. It is at this time when an organ accessible to biopsy is selected (the most accessible after evaluating the imaging studies).13,16,17
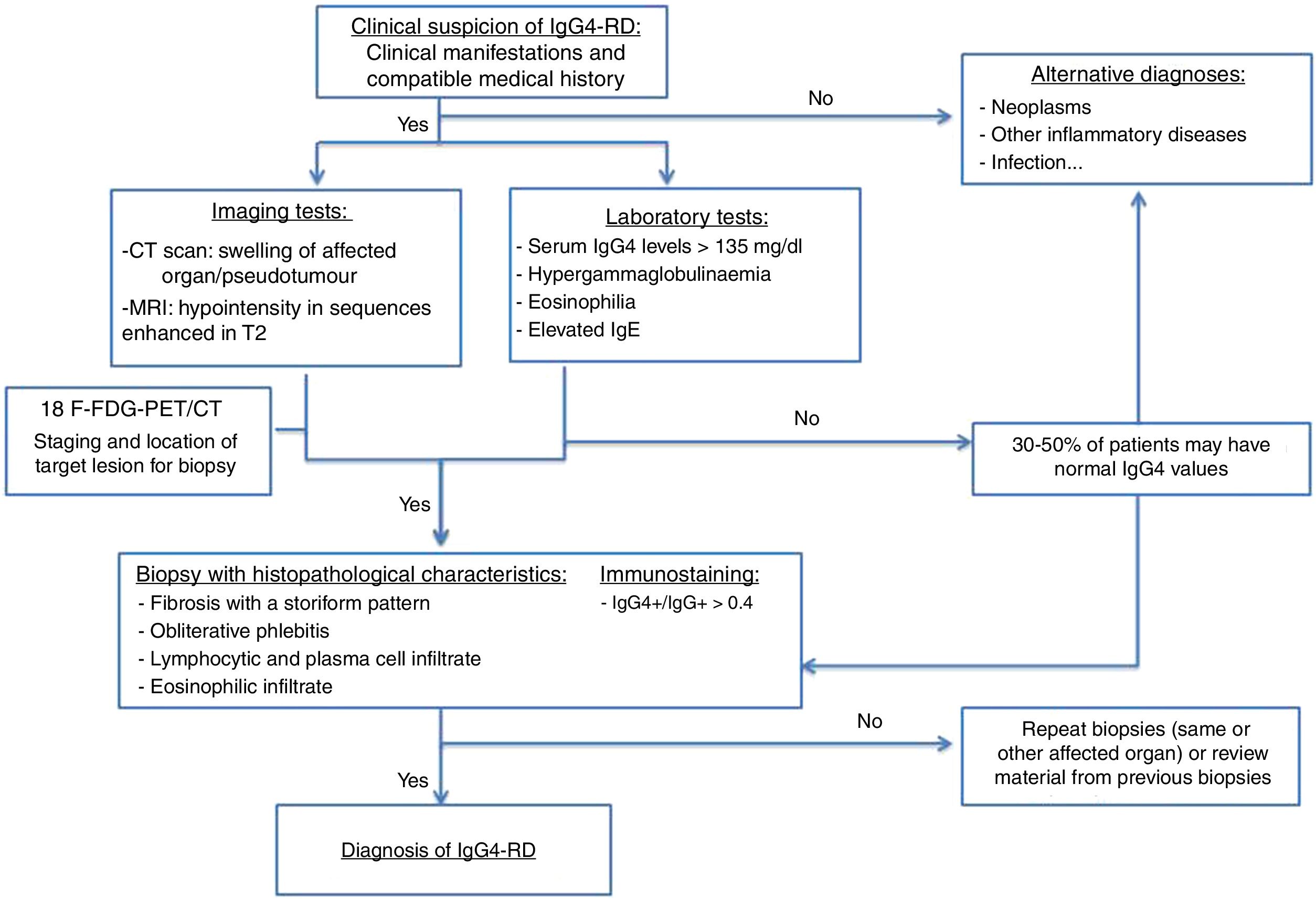
The diagnostic algorithm shown in Fig. 1 is proposed.15,18
Serological tests
Patients with this disease have elevated serum IgG4 levels, with 84 % > 135mg/dl.18,19
However, according to several studies, there is variability in the sensitivity of the elevation of the IGg4 figures, with between 3 and 30 % of patients with the disease having normal plasma figures.
The factors this variability depends on are:
- –
The laboratory means used.
- –
The number of organs affected by the disease.
- –
The patient's geographic origin.
Therefore, elevated plasma levels of IGg4 are neither necessary nor sufficient to diagnose the disease. Elevated IGg4 levels are also found in other conditions, so as a single parameter it is not valid for the diagnosis of the disease. Nevertheless, the degree of elevation does correlate with the number of organs involved.20,21
The monitoring of the serum IgG4 determination is useful in determining activity in some patients, but its measurement is not a single parameter for therapeutic decisions. It has been observed that its concentration drops after corticosteroid treatment in many patients.22
ImmunostainingHigh levels of IgG4-positive (IgG4+) plasma cells in tissues is a distinctive finding of the entity, even when serum IgG4 levels are normal.
It is important to consider the following nuances:
- 1
In IgG4 disease, IgG4+ plasma cells present diffusely throughout the lesion.
- 2
The absolute number of these IgG4+ plasma cells must be interpreted according to the specific tissue. For example, in the AIP the cut-off value is 50 cells per high field; however, in sialadenitis the cut-off value is 100.3
- 3
The IgG4+/IgG+ratio in plasma cells is a more specific tool to diagnose the disease, since many inflammatory entities also raise IgG4 plasma cells, by generally raising the pool of plasma cells.
An IgG4+/IgG+ratio in plasma cells > of 40 % has been proposed as an adequate cut-off value in any organ, although the typical value is 70 % or even higher.4
Histopathological characteristicsHistopathology is currently the key to diagnosis. Although there are no histological criteria established by consensus, the 3 pathological characteristics of IgG4-RD are:
- 1
Lymphoplasmocytic infiltration: infiltration is by lymphocytes and plasma cells. Eosinophils are usually present and neutrophil infiltration is rare. Necrosis and granulomas or xanthogranulomatous changes are atypical, and if present, suggest other diagnoses.
- 2
Storiform fibrosis: fibrosis is a prerequisite for diagnosis, even in patients at the onset of their symptoms. It is characterised by radially arranged collagen fibres, which appear braided on the tissue, and is the unique pattern of this disease. This pattern has a patchy distribution, which may cause a sampling error depending on the material obtained by needle biopsy.3
- 3
Obliterative phlebitis: described as a partial or complete obliteration of medium-sized veins, secondary to lymphoplasmocytic infiltration with obstruction of light. It is a pathognomonic sign of AIP (it appears in 90 % of type 1 and in 57 % of type 2).16
Certain histopathological diagnosis usually requires the presence of 2 of the 3 characteristics described above (usually dense lymphoplasmocytic infiltrate and storiform fibrosis). The histopathological findings described are common to all organs.4
Autoimmune pancreatitisType 1 AIP is the prototype of IgG4-RD. It is estimated to represent 2 % of cases of chronic pancreatitis.7 It is characterised by a periductal infiltrate of IgG4 plasma cells that leads to periductal fibrosis. Over time, parenchymal acini also atrophy, there is loss of lobular architecture and evolution to sclerosis. Clinically, there are no specific symptoms compared to other cases of acute, recurrent or chronic pancreatitis, so patients may present with obstructive jaundice, onset diabetes, steatorrhoea, weight loss, etc.
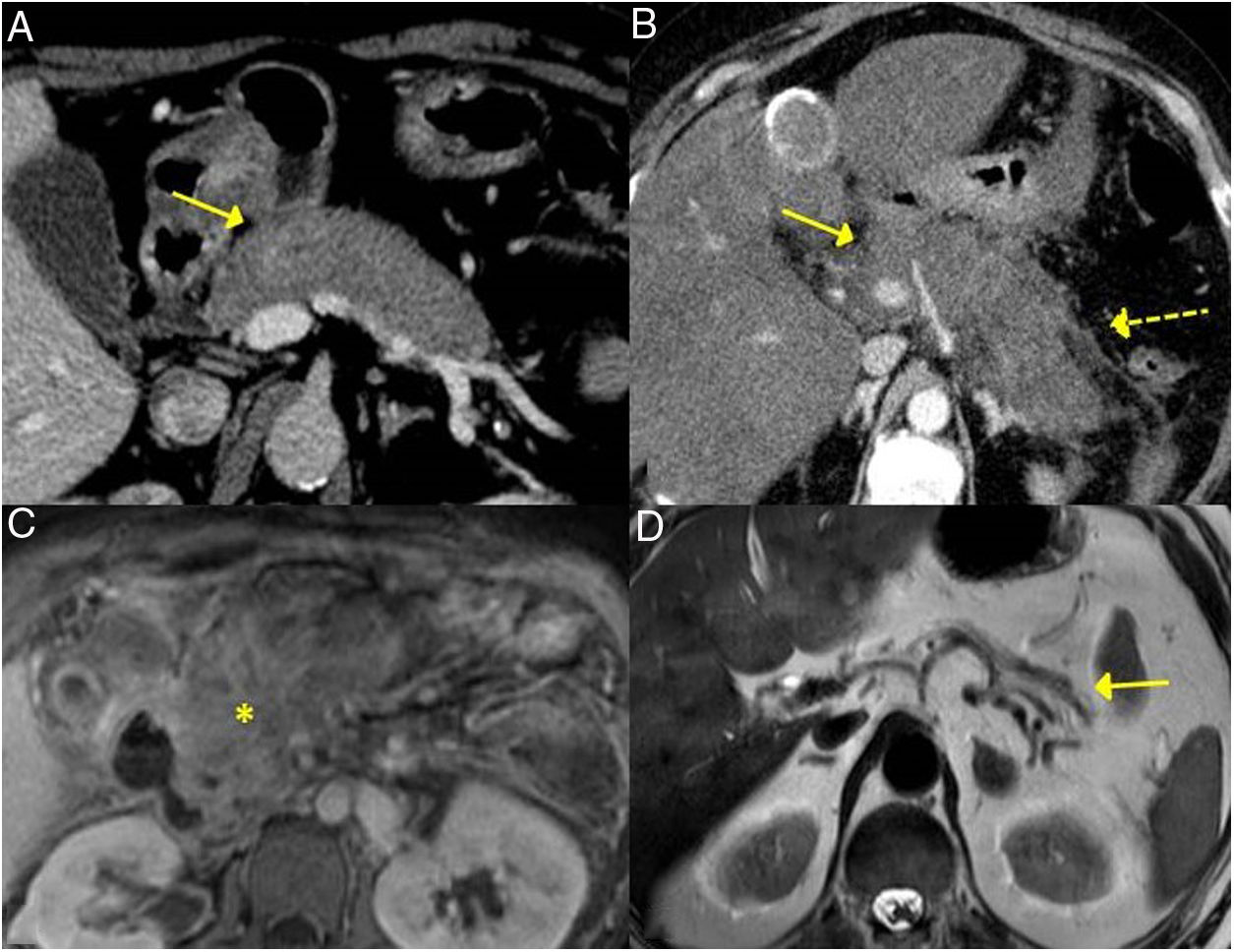
The forms of presentation can be: a picture of obstructive jaundice caused by a mass in the pancreatic head or by thickening of the bile duct wall (up to 55 % of cases), a similar picture to tumour; an increase in pancreas size in imaging tests, diffusely or focally, with irregular narrowing of the pancreatic duct, and sometimes also of the common bile duct; simulating single or recurrent acute pancreatitis and evolving into chronic pancreatitis with calcifications and exocrine and endocrine pancreatic insufficiency17,23 (Fig. 2).
 CT scan image, diffuse increase in pancreatic size (continuous arrow) without associated peripancreatic inflammatory changes. B) CT scan image, diffuse increase in pancreatic size (continuous arrow) with inflammatory changes in the peripancreatic fat (dashed arrow), indistinguishable from acute pancreatitis. C) MRI image, enhanced sequence in T1 with contrast, fibrotic tissue in pancreatic topography (asterisk). D) MRI image, enhanced in T2, severe pancreatic atrophy (continuous arrow) in a patient with endocrine and exocrine pancreatic insufficiency.")
Different forms of presentation and evolution of type 1 AIP. A) CT scan image, diffuse increase in pancreatic size (continuous arrow) without associated peripancreatic inflammatory changes. B) CT scan image, diffuse increase in pancreatic size (continuous arrow) with inflammatory changes in the peripancreatic fat (dashed arrow), indistinguishable from acute pancreatitis. C) MRI image, enhanced sequence in T1 with contrast, fibrotic tissue in pancreatic topography (asterisk). D) MRI image, enhanced in T2, severe pancreatic atrophy (continuous arrow) in a patient with endocrine and exocrine pancreatic insufficiency.
It is common that other fibrous-inflammatory effects are associated and present, for example, with low back pain secondary to retroperitoneal fibrosis or hydronephrosis, such findings contributing to the diagnosis of the entity.24
Bile duct/IgG4-related sclerosing cholangitisBile duct involvement is the second most frequent common AIP. It can appear in as many as 70 % of patients.7 Some 60–80 % of patients with type 1 AIP have involvement of the hepatobiliary system (bile ducts and gallbladder).25 Nevertheless, IgG4 sclerosing cholangitis can also occur without pancreatic alteration, in which case its diagnosis is very difficult.17
It can affect both the intrahepatic and extrahepatic bile ducts, producing a dense infiltrate of IgG4+ plasma cells in the bile ducts and subsequently fibrosis.26,27
These findings are easily seen in endoscopic retrograde cholangiopancreatography or MRI cholangiography (Fig. 3).
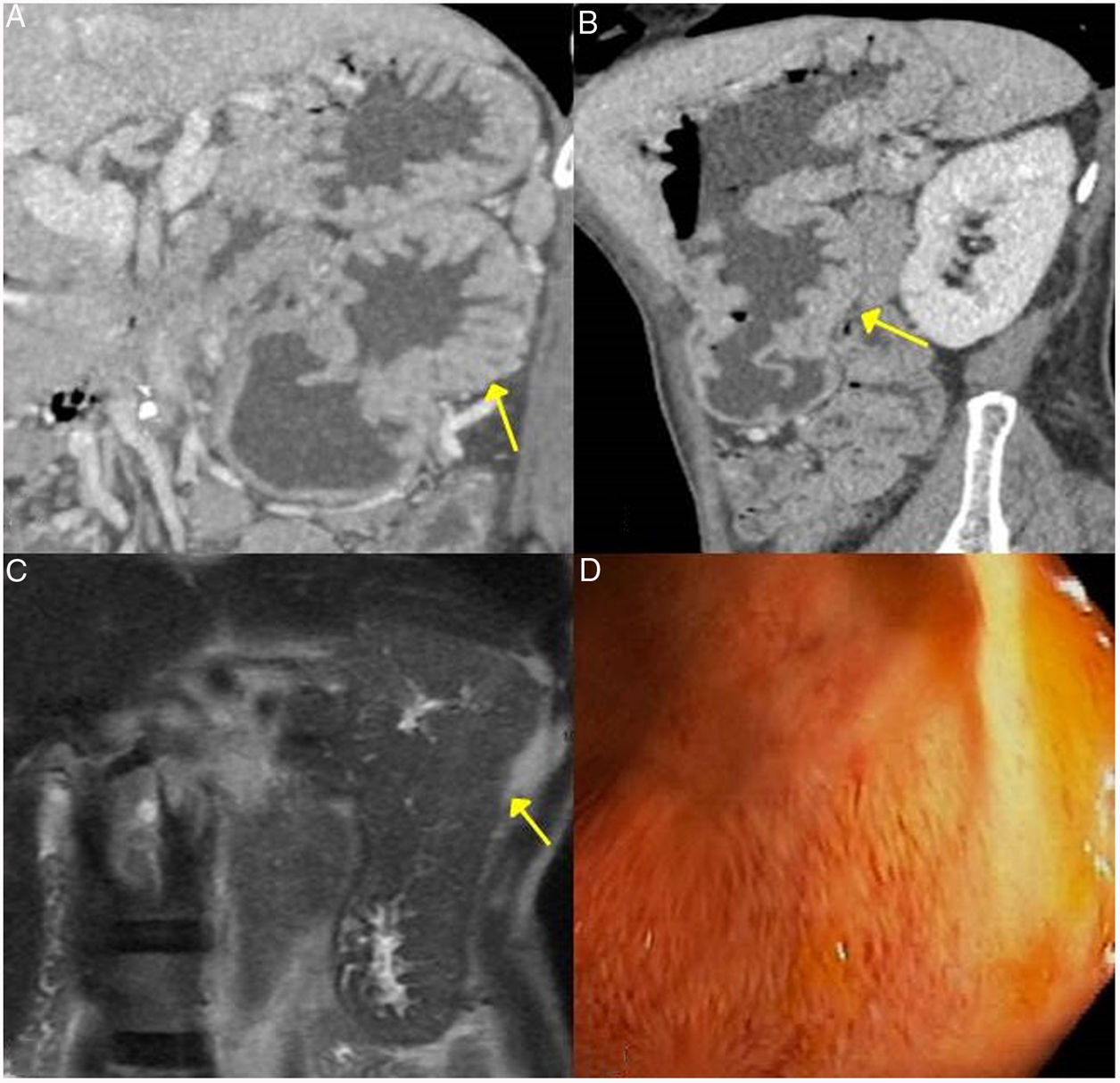
 CT scan images, coronal and sagittal reconstruction respectively: an increase in gastric size with large, thick folds (arrows) is observed. C) MRI image, coronal enhanced sequence in T2: thickening of gastric folds with marked decrease in the gastric lumen is observed. D) Gastroscopy image: mucosal hypertrophy with prominent vascularisation is observed.")
Gastric IgG4-RD. A and B) CT scan images, coronal and sagittal reconstruction respectively: an increase in gastric size with large, thick folds (arrows) is observed. C) MRI image, coronal enhanced sequence in T2: thickening of gastric folds with marked decrease in the gastric lumen is observed. D) Gastroscopy image: mucosal hypertrophy with prominent vascularisation is observed.
The most frequently affected segment is the intrapancreatic portion of the bile duct, because most often this entity coexists with AIP, with thickening of the bile duct wall due to lymphoplasmocytic infiltration occurring alongside inflammation or oedema of the pancreas.17 It induces dilation of the anterograde bile duct with obstructive jaundice. It usually has a more acute clinical presentation and a shorter duration of symptoms. Without treatment it can be self-limiting or evolve to cirrhosis.28 In imaging tests, such as CT and MRI, it may be indistinguishable from cholangiocarcionoma.28–32
The involvement of the gallbladder determines a diffuse thickening of its wall, due to the transmural infiltration of the lymphoplasmocytic infiltrate. The thickening of the wall of the gallbladder appears hypointense in MRI in the images enhanced in T2 and takes up contrast significantly with persistence of the enhancement in the late stages.28
The coexistence of extrabiliary disease, especially pancreatic and renal disease, is highly indicative of IgG4-related cholangitis.
Retroperitoneum and mesenteryInvolvement of the retroperitoneum and mesentery can be in the form of: retroperitoneal fibrosis (most common), periaortitis or sclerosing mesenteritis.14
Retroperitoneal fibrosisIt is estimated that 10 % of patients with AIP develop peritoneal fibrosis. IgG4-related fibrosis has the same imaging findings as secondary retroperitoneal fibrosis.
The most common presentation is like an inflammatory tissue that surrounds the aorta and its branches extending to the retroperitoneum, encompassing the ureters and causing hydronephrosis (sometimes this is the first manifestation) (Fig. 4). The lesion may present as a soft tissue mass simulating malignancy.
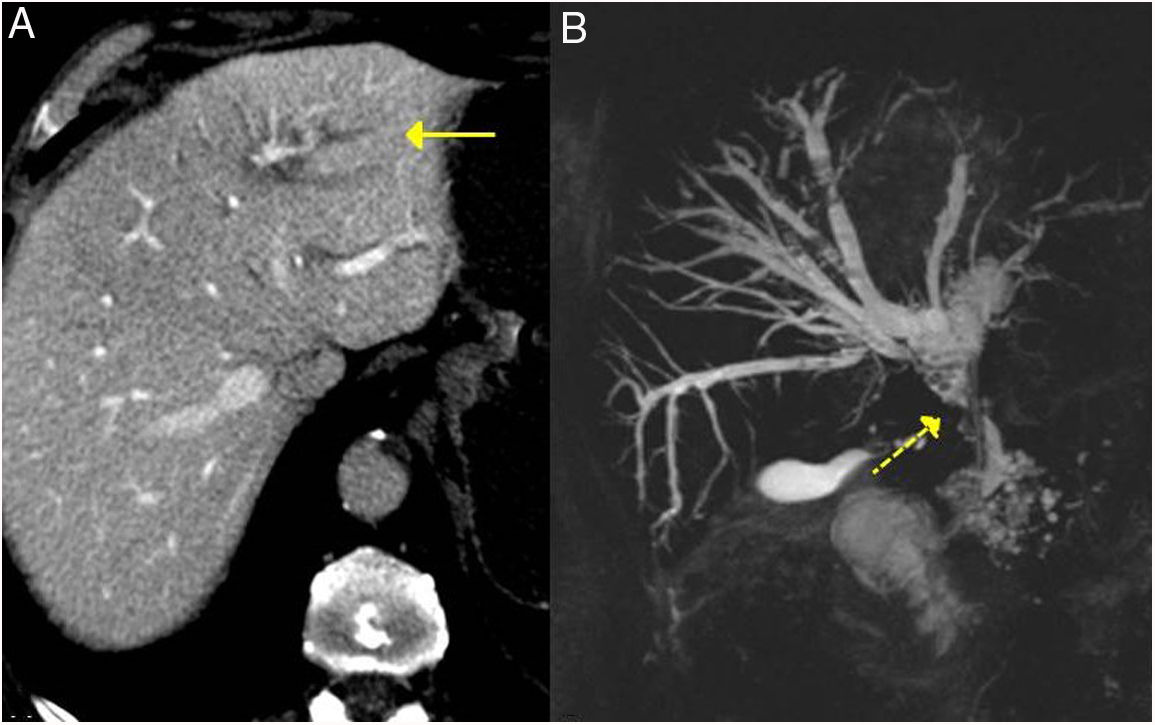
 CT scan image: dilation of the left intrahepatic bile duct is observed (continuous arrow). B) MRI cholangiography image: dilation of the left and right intrahepatic and extrahepatic bile ducts is observed, with stenosis in the middle third of the bile duct (dashed arrow).")
IgG4-related sclerosing cholangitis. A) CT scan image: dilation of the left intrahepatic bile duct is observed (continuous arrow). B) MRI cholangiography image: dilation of the left and right intrahepatic and extrahepatic bile ducts is observed, with stenosis in the middle third of the bile duct (dashed arrow).
The diagnosis is simpler if other lesions coexist (such as pancreatic involvement). It is not uncommon for it to appear in AIP monitoring.33
Many of the retroperitoneal fibrosis cases considered primary that, after the establishment of the IgG4-RD criteria, are now considered as part of this entity, are cases that have a good response to steroid treatment.34
Vascular involvementVascular involvement is usually in the form of periaortitis. The thickening of the vessel wall with enhancement is characteristic. If left untreated, it can evolve into dissection or formation of an aneurysm.
IgG4 involvement is indistinguishable from other forms of non-infectious aortitis, such as those that occur in systemic rheumatic diseases, such as rheumatoid arthritis, Behçet's disease, giant cell arteritis or Takayasu arteritis. Like IgG4 disease, these diseases present with prominent lymphoplasmocytic infiltrate.33
Sclerosing mesenteritisThis is a rare and chronic disorder that encompasses the mesentery of the small intestine with various degrees of lipodystrophy, inflammation, fibrosis and necrosis. The idiopathic disease has been linked to a history of prior abdominal surgery, neoplasms or autoimmune diseases.7 In its variant associated with IgG4-RD, it usually presents as a swollen lesion that encompasses and narrows the mesenteric vessels. This mesenteric mass can occur in contiguity with the AIP lesions and, less frequently, also with sclerosing cholangitis or retroperitoneal fibrosis. It can infiltrate the loops and induce a partial or total intestinal obstruction.17
Digestive tractThe involvement of the gastrointestinal tract in IgG4 disease is rare. There are few cases recorded in the literature and they mostly refer to the stomach and small intestine.35,36
Although infiltration by IgG4+ plasma cells has been described in the gastric and colonic mucosa, and the major papilla in some patients with AIP, until 2013 there was some debate over whether these were lesions related to IgG4-RD or whether IgG4-related gastrointestinal disease could be considered a separate entity, since such cases did not always meet the criteria of dense fibrosis or obliterative phlebitis.37
However, its existence has now been confirmed, despite the fact that the series of cases collected are very scarce. Lim et al. reported 15 cases of gastric involvement in 2018 (of these, 11 with isolated involvement and 4 with coexistent AIP).22
StomachThe different gastric manifestations described are:
- –
Diffuse infiltration in the gastric mucosa by IgG4+ plasma cells, without the other histological criteria of the disease; related to some cases of AIP and that disappears after corticosteroid treatment. Occasionally, this infiltration has led to the formation of nodules in the gastric mucosa of up to 15 mm.37
- –
Focal polypoid lesions or focal masses, up to 3cm, that do meet all the criteria.
- –
Gastric ulcer.
- –
Diffuse thickening of the wall (Fig. 5).
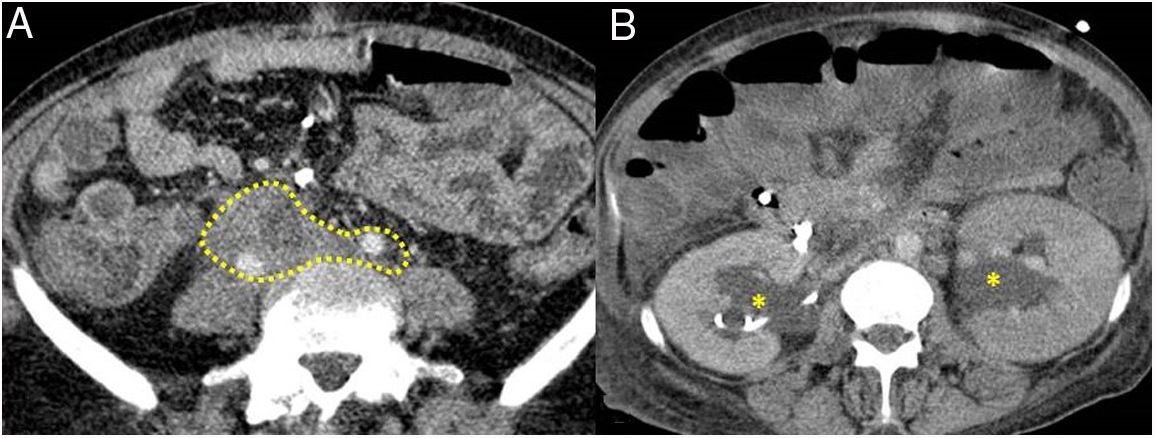
 Dense soft tissues (within the dotted line) surrounding the retroperitoneal structures, such as both ureters and the common iliac arteries, corresponding to retroperitoneal fibrosis. B) Same patient as in A in whom said fibrosis has caused bilateral proximal hydronephrosis (asterisks).") Figure 5.
Figure 5.Retroperitoneal fibrosis. CT scan images. A) Dense soft tissues (within the dotted line) surrounding the retroperitoneal structures, such as both ureters and the common iliac arteries, corresponding to retroperitoneal fibrosis. B) Same patient as in A in whom said fibrosis has caused bilateral proximal hydronephrosis (asterisks).
(0.1MB). - –
Association with local adenopathies.
 Dense soft tissues (within the dotted line) surrounding the retroperitoneal structures, such as both ureters and the common iliac arteries, corresponding to retroperitoneal fibrosis. B) Same patient as in A in whom said fibrosis has caused bilateral proximal hydronephrosis (asterisks).")
These findings are often indistinguishable by imaging from malignant gastric disease (especially primary gastric carcinoma or gastric lymphoma).37–40
OesophagusCases of IgG4-related esophagitis have been described in which a thickening of the oesophageal wall has been observed that caused dysphagia and weight loss, with the disease's own characteristics being demonstrated in the biopsy.37,39
Major duodenal papillaThis is affected in 41–61 % of patients with AIP, with swelling and IgG4+ lymphoplasmocitic infiltrate, findings that also reverse after steroid treatment.41 Therefore, its biopsy and study with immunostaining is an interesting tool to diagnose AIP.
Colonic lesionsAs in the stomach, occasionally in patients with AIP an infiltration by IgG4+ plasma cells is found in the colonic mucosa, but without dense fibrosis or obliterative phlebitis.
Colonic polyposis has also been described in patients with AIP and with marked reduction of polyps after steroid treatment.
Other forms of manifestation of IgG4-RD are as sclerous and circumscribed nodular lesions in the caecum and sigmoid colon, with abundant infiltration by IgG4+ plasma cells, without disease in other organs.37
Conditions not related to IgG4-RD that present with an increase in IgG4-positive cells- –
Inflammatory conditions: oral inflammatory diseases, primary sclerosing cholangitis, rheumatoid arthritis.
- –
Lymphoma: Low-grade B-cell lymphomas should be excluded in cases of possible IgG4-RD with florid lymphoplasmocytic infiltrates. Those that simulate this entity are usually mainly extranodal marginal zone lymphoma and sometimes follicular and angioimmunoblastic lymphomas. Finding CD20+ cells and restriction of immunoglobulin light chains, as well as aggregates of B lymphocytes, supports the diagnosis of lymphoma.
- –
Malignant disease: the cancerous tissue may be infiltrated with IgG4+ plasma cells to varying degrees. It is more common in pancreatobiliary cancers, although it can also be seen in other tumours. IgG4 infiltration in malignant tissues is usually patchy and not associated with the rest of the characteristics of the disease, such as the storiform pattern or phlebitis.4
It is important to start treatment as soon as possible once the disease is diagnosed, in order to prevent the development of fibrosis, and to maintain the treatment once remission is achieved to avoid relapses. IgG4-RD is mainly treated with systemic glucocorticoids. Although there are studies with proposals for the treatment of the disease, there is no established comprehensive management of it. In part, this is because the clinical symptoms depend on the organs involved, and the current consensus is based on the treatment of type 1 AIP, as a representative entity of the disease.42
Induction of remissionInduction of remission is successfully achieved with steroids in up to 90 % of patients with type 1 AIP (Fig. 6). Both the Japanese guidelines and the international consensus propose steroids as the first line of treatment, including in asymptomatic patients who present with an image of persistent pancreatic mass or who present with IgG4-sclerosing cholangitis with abnormal liver function in serological tests. When there are contraindications to steroids, rituximab can induce remission as a sole treatment as well.1,43
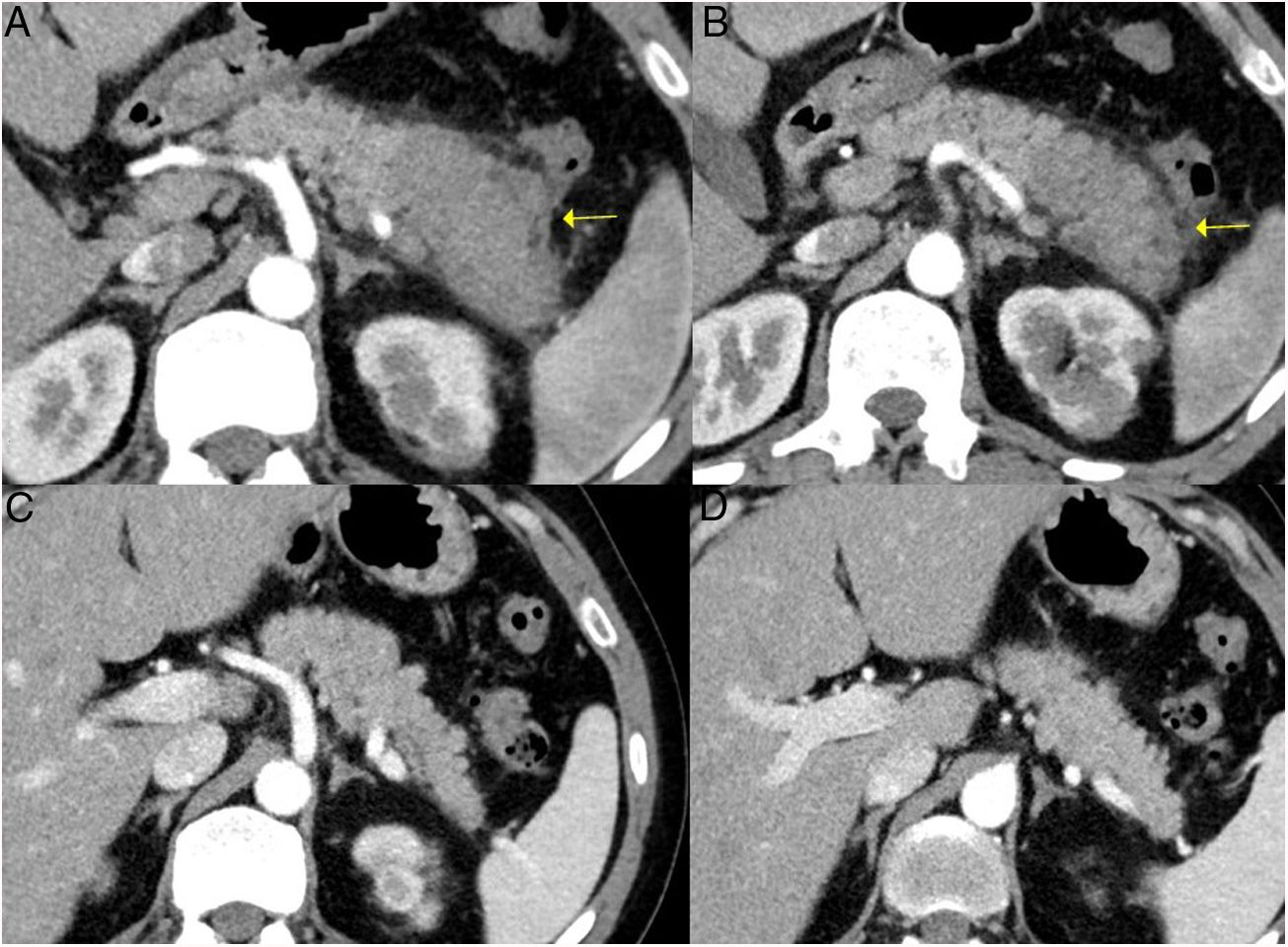
 CT scan images of a patient with focal pancreatitis of the pancreatic tail (arrows) with increased focal size, loss of pancreatic clefts and characteristic peripheral hypodense halo, in the context of an IgG4-RD. C and D) CT scan images at 3 months after the patient received corticosteroid treatment: complete resolution of the focal involvement of the pancreatic tail is observed, which is shown to be of normal size and with normal pancreatic clefts preserved.")
Remission of focal type 1 AIP of the pancreatic tail after corticosteroid treatment. A and B) CT scan images of a patient with focal pancreatitis of the pancreatic tail (arrows) with increased focal size, loss of pancreatic clefts and characteristic peripheral hypodense halo, in the context of an IgG4-RD. C and D) CT scan images at 3 months after the patient received corticosteroid treatment: complete resolution of the focal involvement of the pancreatic tail is observed, which is shown to be of normal size and with normal pancreatic clefts preserved.
Relapses of type 1 AIP occur in up to 30–50 % of cases. Japanese studies suggest that relapses are related to diffuse involvement of the pancreas, with plasma levels of IgG4 that are elevated or that reduce little with treatment, as well as the association of type 1 AIP with IgG4-sclerosing cholangitis, especially when it is of the proximal type In addition, they propose these findings as early predictors of relapse.
In relapses, initial treatment with corticosteroids is still useful, although there is less consensus. The international consensus proposes re-administering corticosteroids, increasing their dose or adding immunosuppressive agents or rituximab.1,43
Maintenance treatmentMaintenance therapy consists of low doses of corticosteroids, with studies (mostly Asian) suggesting this monotherapy for prevention of relapses. Other alternatives for maintenance treatment are rituximab (with different therapeutic strategies in terms of dose and duration of maintenance treatment)44–46 and immunomodulators such as azathioprine, 6-mercaptopurine or mycophenolate mofetil. In cases with focal pancreatic alteration, low plasma IgG4 levels, without extrapancreatic disease and with resolution of the findings on imaging after induction treatment, maintenance treatment would not be indicated.42,43 In cases of severe organ damage, surgery or radiation therapy may be necessary.42
PrognosisAlthough remission is common in cases of type 1 AIP, with resolution of symptoms, long-term evolution is not well studied. Some 10 % progress to chronic pancreatitis with calcifications and exocrine insufficiency.
The risk factors associated with relapses are:
- –
Obstructive jaundice.
- –
Extrapancreatic biliary involvement.
- –
Intra- or extrahepatic proximal ductal stenosis.
- –
Incomplete remission of radiological or serological findings during the maintenance period.
- –
Diffuse increase of the baseline pancreatic volume.7
There appears to be an increased risk of cancer (gastric, lung, prostate, colon, non-Hodgkin lymphoma, bile duct and thyroid) in patients with IgG4-related pancreatitis, especially in the year after diagnosis, which would indicate that in some patients this disease could be a paraneoplastic syndrome. The presence of significant k-ras mutations has been demonstrated in samples of pancreas and bile duct tissue from patients with AIP, which indicates the possibility that this entity is a risk factor for the development of pancreatic and biliary tumours.47
ConclusionsIgG4-RD can affect multiple organs simultaneously or at different periods. Its clinical manifestations will depend on the affected organs and can simulate other diseases such as neoplasms. It is necessary to know of this disease, suspect it and make an appropriate diagnosis, thereby avoiding unnecessary surgeries and the development of irreversible fibrosis with the consequent severe organ failures.
Conflict of interestsThe authors declare that they have no conflicts of interest.
Please cite this article as: Sánchez-Oro R, Alonso-Muñoz EM, Martí Romero L. Revisión de la enfermedad relacionada con la IgG4. Gastroenterol Hepatol. 2019;42:638–647.
