




INTRODUCCIÓN
El síndrome de Cronkhite-Canada es una poliposis, sin agregación familiar y caracterizada por la presencia de pólipos gastrointestinales no adenomatosos y alteraciones ectodérmicas. A pesar de estar considerada como una poliposis no maligna, el pronóstico en general es malo, y actualmente no cuenta con un tratamiento etiológico efectivo.
OBSERVACIÓN CLÍNICA
Varón de 58 años de edad, sin antecedentes familiares o personales de interés, salvo tabaquismo sin criterios clínicos de bronquitis crónica. Desde hacía 4 meses refería hipoageusia, malestar epigástrico que se aliviaba con el vómito, diarrea sin productos patológicos y una pérdida ponderal de 10 kg de peso.
A la exploración, el paciente presentaba mal estado nutricional, disminución de la masa muscular, pérdida del vello corporal y del cuero cabelludo, onicopatía, máculas parduscas en las palmas de las manos y edemas perimaleolares. No se palpaban adenopatías. La auscultación cardiopulmonar era normal. El abdomen era blando, depresible y no doloroso a la palpación, sin masas o megalias, con peristaltismo presente.
En la analítica, el hemograma, la coagulación, la glucosa, los iones, la función renal, el perfil hepático y lipídico se obtuvieron valores dentro de la normalidad. El paciente presentaba hipoproteinemia (4,6 g/dl), hipoalbuminemia (2,26 g/dl), sin proteinuria y una alfa-1-antitripsina fecal elevada (3,49 mg/g; valor normal, < 0,3 mg/g). El coprocultivo, los parásitos y la grasa en heces, los anticuerpos antiendomisio y antireticulina, así como el test de la D-xilosa y los marcadores tumorales, eran negativos. La determinación de cinc sérico fue de 42 µg/dl (60-150 µg/dl).
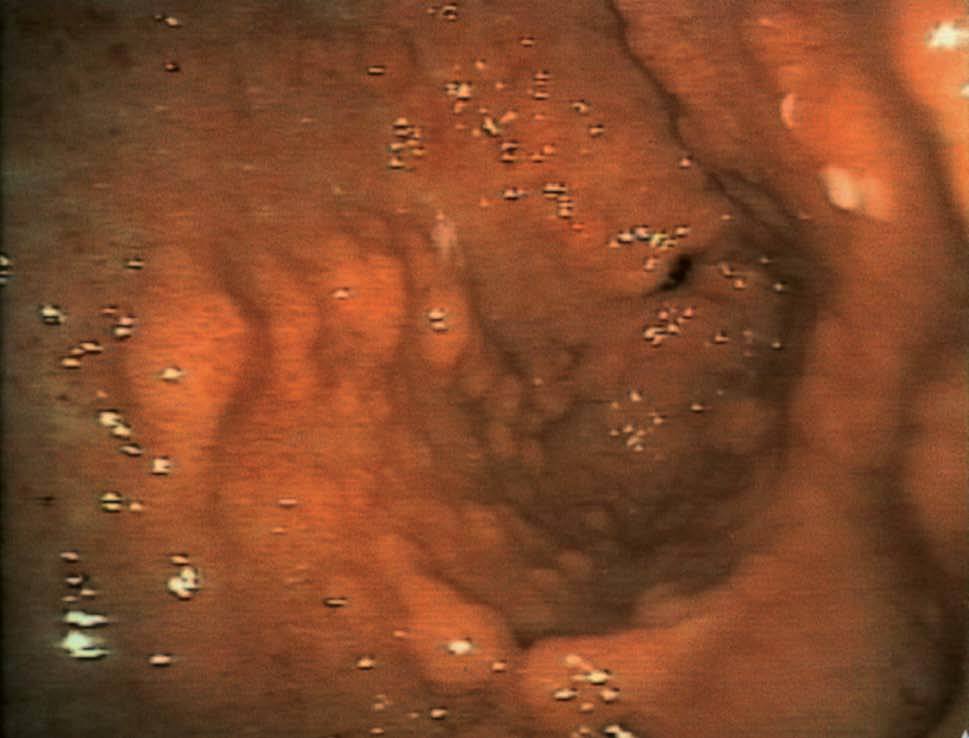
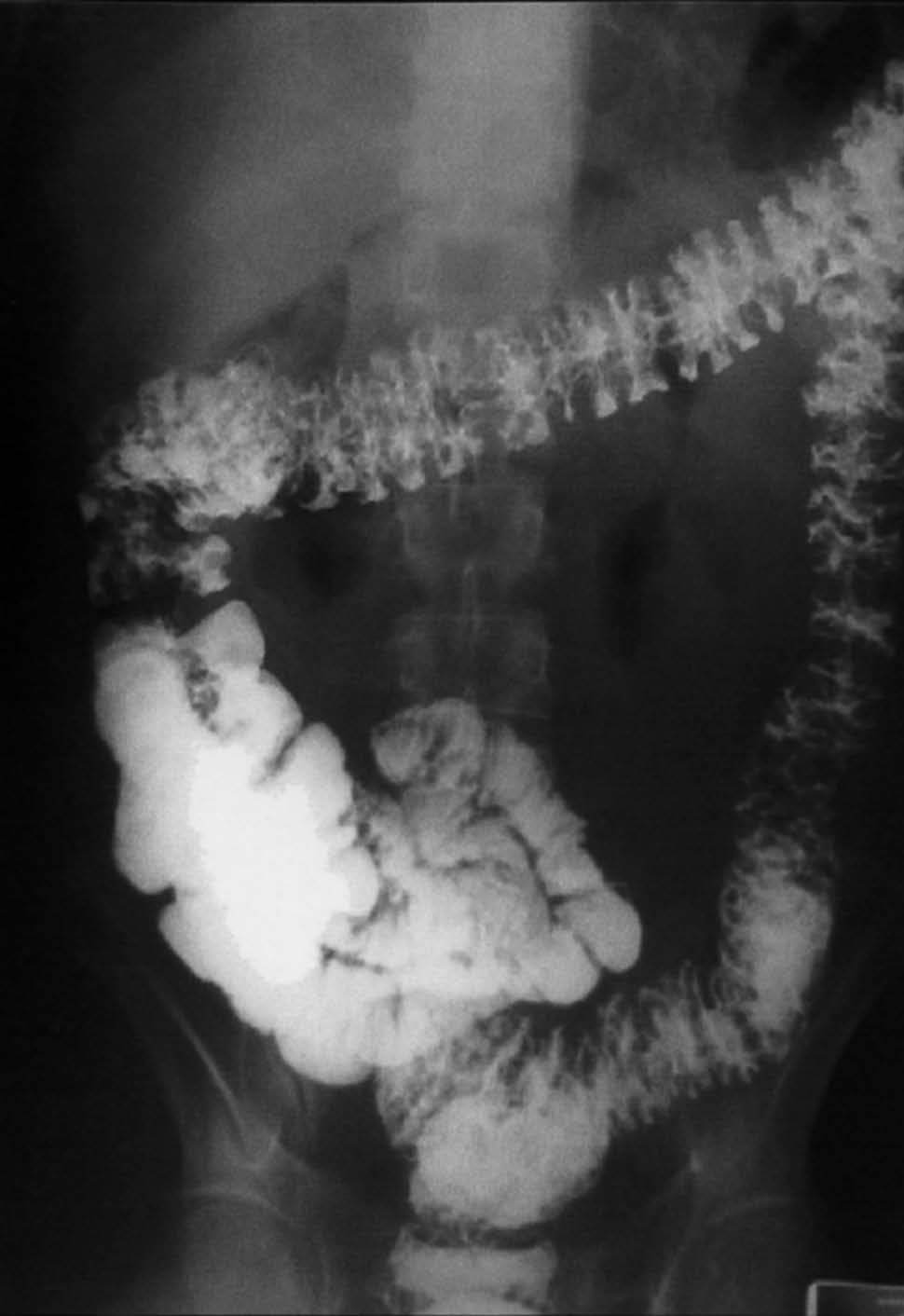
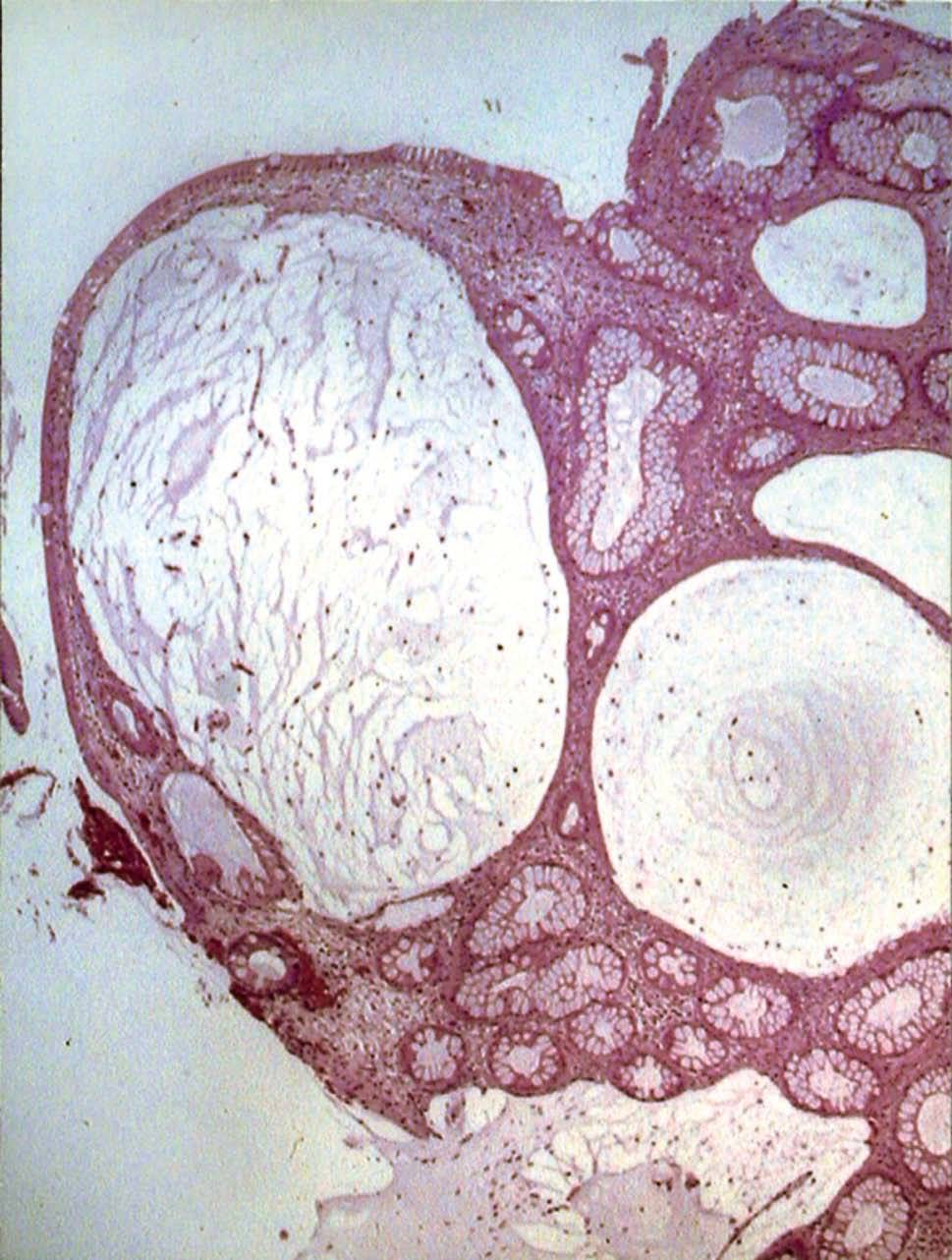
Se realizó una endoscopia oral, que mostró un esófago normal y una cámara gástrica con múltiples pólipos sésiles, de 3-8 mm de diámetro, de superficie lisa y eritematosa, algunos erosionados (fig. 1). En el bulbo y la segunda porción duodenal había pólipos de menor tamaño. El test de la ureasa de la mucosa antral fue positivo. Se tomaron muestras para el estudio histológico en el antro, la primera y la segunda porción duodenal. En el tránsito gastrointestinal se hallaron nódulos de 4 mm de diámetro en la tercera porción duodenal y el yeyuno proximal, así como una aceleración del tránsito y la fragmentación de la columna de bario. La ecoendoscopia definió una mucosa gástrica engrosada (7 mm) con dilataciones quísticas superficiales. En una rectosigmoidoscopia, realizada hasta 40 cm del margen anal, se apreciaron desde la unión rectosigmoidea múltiples pólipos, que no superaban los 6 mm de diámetro, sésiles y de superficie brillante, eritematosa y lisa. Se practicó una polipectomía endoscópica con asa de alambre de uno de los pólipos para su estudio histológico. En el enema opaco se apreciaban imágenes nodulares en todo el marco cólico, de predominio en el colon descendente y el sigma (fig. 2). Una tomografía computarizada abdominopélvica definió un engrosamiento difuso de los pliegues gástricos y de las asas intestinales, sin adenopatías locorregionales. El estudio histológico de las muestras obtenidas, tanto del tracto digestivo superior como del colon, puso de manifiesto una mucosa con dilataciones quísticas glandulares y agregados de polimorfonucleares en su interior, así como un edema de la estroma y un infiltrado inflamatorio linfoplasmocitario (fig. 3).
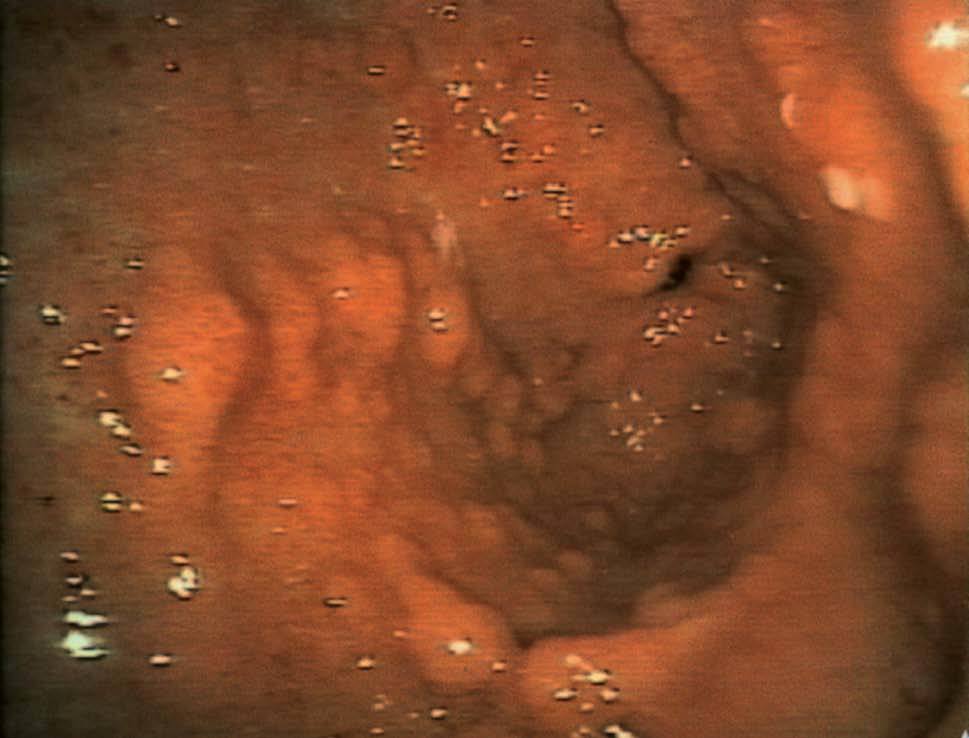
Fig. 1. Endoscopia oral.
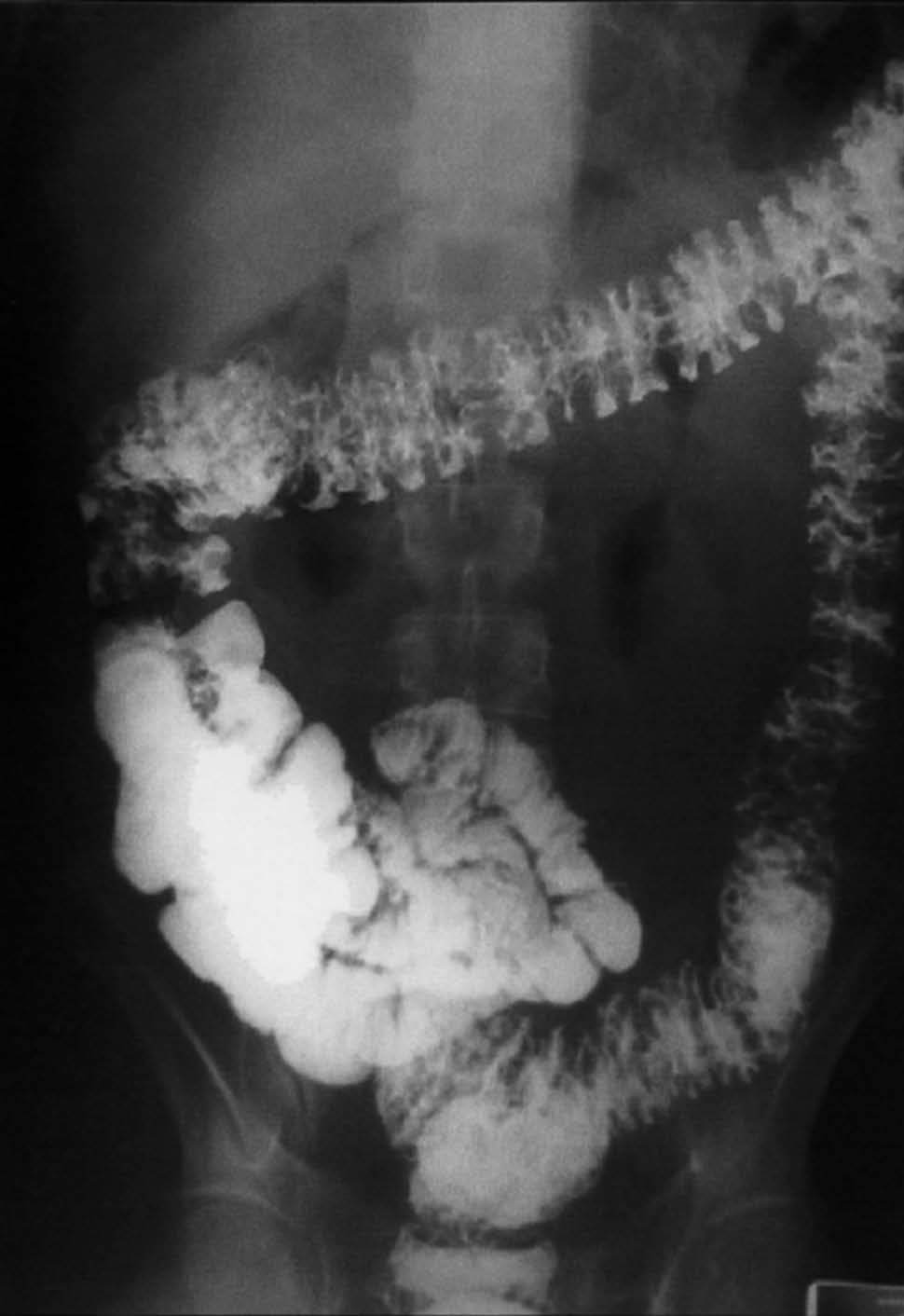
Fig. 2. Enema opaco con imágenes nodulares en todo el marco cólico, de predominio en el colon descendente y el sigma.
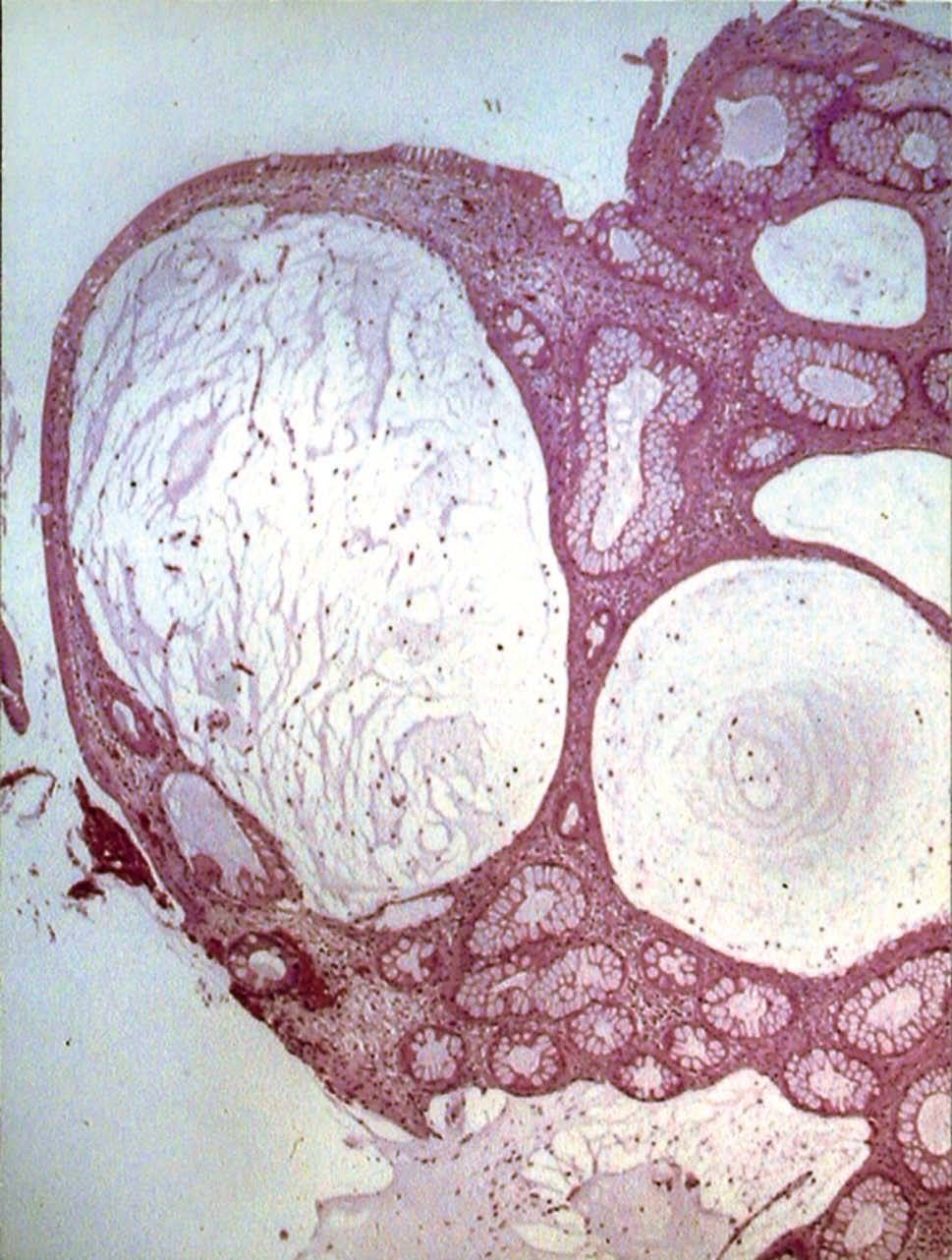
Fig. 3. Estudio histológico de las muestras obtenidas, que puso de manifiesto una mucosa con dilataciones quísticas glandulares y agregados de polimorfonucleares en su interior, así como un edema de la estroma y un infiltrado inflamatorio linfoplasmocitario.
Se administró soporte nutricional oral, erradicación de Helicobacter pylori (omeprazol 20 mg/12 h, amoxicilina 1 g/12 h y claritromicina 500 mg/12 h) y 300 mg de sulindaco/día, con lo que se obtuvo una mejoría clínica y analítica de corta duración. Se inició entonces tratamiento intravenoso durante una semana con 40 mg de metilprednisolona (0,8 mg/kg/día), cromoglicato disódico (200 mg/6 h), ranitidina (300 mg/día), loratadina (10 mg/día), ciprofloxacino (500 mg/12 h) y suplementos con cinc. Posteriormente, se introdujeron corticoides po vía oral, reduciendo 4 mg de metilprednisolona cada semana y manteniendo el resto de la medicación. Se obtuvo una remisión clínica precoz y el paciente recuperó el sentido del gusto, el vello corporal y del cuero cabelludo y 14 kg de peso. Tras finalizar el tratamiento corticoideo, administrado durante un total de 10 semanas, se realizó al paciente una endoscopia oral, en la que se observó que la mayor parte de las lesiones polipoides gástricas y duodenales habían desaparecido. Persistía únicamente una mucosa gástrica nodular en la cara anterior del cuerpo gástrico. El paciente ha continuado tratándose con cromoglicato disódico, ranitidina y loratadina, y en la actualidad se mantiene en remisión clínica, 6 meses después del inicio del tratamiento.
DISCUSIÓN
El síndrome de Cronkhite-Canada es una entidad infrecuente descrita por primera vez en 19551. Se trata de una poliposis no adenomatosa y no hereditaria que afecta difusamente al tracto gastrointestinal y asocia anormalidades ectodérmicas: alopecia, hiperpigmentación y onicopatía.
Aunque se han publicado casos en pacientes caucasianos, este síndrome es más prevalente en Asia2, suele iniciarse en la sexta década de la vida3 y presenta una ligera predilección por el sexo masculino4.
Su etiopatogenia permanece en la actualidad sin aclarar. Se han sugerido diversas causas posibles: sobrecrecimiento bacteriano, déficit inmunitario y malabsorción5, deficiencias nutricionales, alteración en la producción de mucina intestinal6 o disfunción mastocitaria7.
Los síntomas más comunes son diarrea, pérdida de peso, náuseas, vómitos, hipoageusia y anorexia. En la exploración física se identifican usualmente alteraciones ectodérmicas, como distrofia ungueal, alopecia del cuero cabelludo y corporal e hiperpigmentación cutánea en las palmas, las plantas, el cuello y la cara7, en forma de máculas pardas por depósitos de melanina en la capa basal2.
Los pólipos son hamartomas, semejantes a los que aparecen en la poliposis juvenil2, que se distribuyen de forma difusa a lo largo del tracto gastrointestinal sin afectar al esófago. Estos pólipos suelen ser sésiles y con erosiones superficiales8. El examen microscópico revela una dilatación quística glandular con moco y agregados de polimorfonucleares, junto a edema de la lámina propia e infiltrado inflamatorio linfoplasmocitario7.
Aunque generalmente se considera que este tipo de pólipos carece de potencial maligno, se han hallado cambios adenomatosos9 y/o carcinomas en el estómago5, el colon10,11 y el recto12 en pacientes con síndrome de Cronkhite-Canada. Hasta el día de hoy se han publicado 21 casos de neoplasias malignas digestivas en pacientes diagnosticados de síndrome de Cronkhite-Canada; sin embargo, se desconoce si estos casos son hallazgos casuales o el resultado del curso evolutivo de la enfermedad.
La falta de una terapia efectiva, su curso progresivo y las complicaciones asociadas definen un mal pronóstico a corto plazo, con una mortalidad de hasta el 60%6. Esta mortalidad se produce fundamentalmente como consecuencia de la desnutrición, si bien se han referido otras complicaciones, como el sangrado gastrointestinal, la intususcepción y la perforación colónica.
El síndrome de Cronkhite-Canada es una enfermedad excepcional en nuestro medio, que plantea el diagnóstico diferencial con otras poliposis difusas no adenomatosas y no hereditarias, como la poliposis lipomatosa, la poliposis linfoide múltiple, la poliposis hiperplásica difusa, los pólipos inflamatorios difusos en relación con parasitosis y/o tuberculosis intestinal, y la hiperplasia nodular linfoide no asociada a inmunodeficiencias primarias7.
Se han ensayado tratamientos tan dispares como los soportes nutricionales orales, la nutrición parenteral total, la combinación de distintos antibióticos6, corticoides13, cinc14, ranitidina15 y cromoglicato disódico, con los que se han obtenido algunas remisiones parciales y/o completas de corta duración16. Recientemente, se ha publicado un caso en el que obtuvo la curación clínica y endoscópica mediante la utilización combinada de prednisona, cromoglicato disódico, ranitidina, loratadina y ciprofloxacino17. Aunque se han comunicado resecciones quirúrgicas del segmento afectado ante determinadas complicaciones, como hemicolectomía en un caso de enteropatía pierdeproteínas, al tratarse de una entidad que afecta difusamente al tracto gastrointestinal, el tratamiento quirúrgico no suele estar indicado3.
En nuestro caso, con la administración combinada de corticoides, ranitidina, cromoglicato disódico, loratadina, ciprofloxacino y suplementos con cinc, al igual que en el caso publicado por Ward et al16, obtuvimos la remisión clínica y endoscópica parcial.
Aunque desconocemos la etiopatogenia y el tratamiento efectivo que mejore el pronóstico de los pacientes con síndrome de Cronkhite-Canada, destacamos que esta poliposis adquirida puede revertirse con tratamiento médico.

