






To review the effectiveness and safety of switching from an originator anti-TNF (Remicade®) to a biosimilar (CT-P13) in patients with inflammatory bowel disease (IBD).
MethodsElectronic and manual search up to September 2017.
ResultsWe identified 24 studies evaluating switching between Remicade® and CT-P13 in 1326 patients. Disease control (no worsening after switching) was confirmed in most of the patients (weighted mean, 88%; 95% CI=86–89%). No unexpected adverse effects were reported in any of the studies.
ConclusionThe risks of switching from Remicade® to a biosimilar seem to be purely theoretical and are not supported by the (still limited) real-world clinical practice experience. On the contrary, a steadily increasing number of publications have shown that there seem to be no safety or efficacy concerns about switching. Therefore, switching from originator to biosimilar infliximab in patients with IBD may be considered acceptable.
Revisar la efectividad y la seguridad del cambio entre anti-TNF original (Remicade®) y biosimilar (CT-P13) en pacientes con enfermedad inflamatoria intestinal (EII).
MétodosBúsqueda electrónica/manual hasta septiembre de 2017.
ResultadosSe identificaron 24 estudios que evaluaban el switching entre Remicade® y CT-P13 en 1.326 pacientes. Se confirmó el control de la enfermedad (es decir, la ausencia de empeoramiento clínico tras el switching) en la mayoría de los pacientes (media ponderada: 88%; IC 95%: 86-89%). No se describieron efectos adversos inesperados en ninguno de los estudios.
ConclusiónEl riesgo de cambiar de Remicade® a un biosimilar parece ser meramente teórico y no estar sustentado por la —aún limitada— experiencia de práctica clínica en la vida real. Por el contrario, el progresivamente creciente número de publicaciones indica que no parece haber motivo de preocupación en términos de eficacia ni seguridad. Por tanto, el switching entre infliximab original y biosimilar en pacientes con EII puede considerarse aceptable.
Biological medicinal products (or biologics) are active substances derived from living cells or organisms with the aid of biotechnology methods (recombinant DNA, controlled gene expression, antibody technologies).1 The cytokine tumor necrosis factor (TNF) α is a key mediator of inflammation in inflammatory bowel disease (IBD). Therefore, the use of anti-TNF drugs, which target TNF, has greatly advanced the armamentarium for the treatment of both Crohn's disease (CD) and ulcerative colitis (UC). Infliximab, adalimumab, golimumab and certolizumab have shown significant efficacy in IBD patients,2 being associated with relevant clinical benefits such as mucosal healing, fewer hospitalizations and surgical needs, and improved quality of life.3–6
Although biologic agents have revolutionized the care management of IBD and many other life-threatening and debilitating diseases, the long-term use of these agents may be very expensive, placing a significant burden on National Healthcare Systems. Recent or impending expiry of patents for some key biologics, such as Remicade® (infliximab) in the case of IBD, has led to development of biosimilar products. A biosimilar is a biological medicinal product that contains a version of the active substance of an already authorized original biological medicinal product.7 A biosimilar establishes similarity to the reference medicinal product in terms of quality characteristics, biological activity, safety, and efficacy based on a comprehensive comparability exercise.8–11
Biosimilar development has been set out to accomplish two main goals: to reduce the expenditure of healthcare on costly biological treatments and to improve patient's access to these drugs. Biosimilars of infliximab (CT-P13) were first approved by the European Medicine Agency (EMA) in 2013. Initially, the European Crohn's Colitis Organisation raised some caution on the use of biosimilars.12 An insufficient understanding of the characteristics and use of biosimilars became evident in a web survey performed among European Crohn's and Colitis Organisation members in the early phases.13 However, since biosimilars were introduced in the European Union (EU) market in early 2015, more data from IBD patients have supported the biosimilarity of biosimilar infliximab CT-P13 and the reference product, with no significant differences in terms of efficacy or safety.14 Consideration of these findings, together with a better understanding of the process of biosimilar development and regulatory approval, have contributed to a change in the perception of IBD experts, who now prescribe biosimilars with significantly more confidence.15
Nonetheless, an important unanswered question for prescribing physicians is whether it is possible to switch from reference medicinal product (Remicade®) to biosimilar (CT-P13) in patients with IBD without any detrimental effects on safety and efficacy. This fact has generated an intensive debate about the issue of switching, and some national societies have expressed concerns. Furthermore, many physicians are reluctant to switch from Remicade® to CT-P13 in view of the limited evidence on relevant clinical outcomes, specifically in IBD patients, and the issue of immunogenicity is of significant concern.
Our aim is to perform a critical review on the data evaluating the effectiveness and the safety of switching from reference medicinal products to biosimilars, specifically focusing on the experience of switching between Remicade® and CT-P13 in patients with IBD. The index of contents of the present review is summarized in Table 1.
Index of contents of the review.
| 1. Biosimilar definition. |
| 2. Interchangeability between originator and biosimilar. |
| 3. Switching between biologicals different from anti-TNFs. |
| 4. Switching between Remicade® and CT-P13 in diseases different from inflammatory bowel disease. |
| 5. Switching between Remicade® and CT-P13 in inflammatory bowel disease: efficacy. |
| 6. Switching between remicade® and CT-P13 in inflammatory bowel disease: safety and immunogenicity. |
| 7. Methodological limitations of switching studies in inflammatory bowel disease. |
| 8. The NOR-SWITCH trial. |
| 9. Switching in inflammatory bowel disease: ongoing studies. |
| 10. Cost saving of switching in inflammatory bowel disease. |
| 11. Recommendations of health and scientific organizations on switching in inflammatory bowel disease. |
| 12. The opinion of the patient. |
| 13. Conclusions. |
A systematic bibliographic search was designed to identify studies investigating switching from reference medicinal product (Remicade®) to biosimilar (CT-P13) in patients with IBD (CD or UC). An electronic search was performed in PubMed up to September 2017 using the algorithm summarized in Fig. 1. International conference abstracts were inspected by means of a manual search of American Digestive Disease Week, United European Gastroenterology Week, and the conferences of the European Crohn's and Colitis Organisation. In addition, the reference lists from the selected articles were reviewed to identify additional studies of potential interest. Articles published in any language were included. If a study was duplicated, the most recent one fulfilling the inclusion criteria was included.
Biosimilar definition
The EMA defines a biosimilar as a biological medicinal product that contains a version of the active substance of an already authorized original biological medicinal product (reference medicinal product).7 The US Food & Drug Administration (FDA) defines a biosimilar as a biological product that is highly similar to the reference product notwithstanding minor differences in clinically inactive components.16 There are no clinically meaningful differences between the biological product and the reference product in terms of the safety, purity, and potency of the product.17 Finally, the World Health Organization (WHO) defines a biosimilar as a biotherapeutic product which is similar in terms of quality, safety and efficacy to an already licensed reference biotherapeutic product.18
Thus, in accordance with regulatory frameworks laid out by the EMA, the FDA, the WHO and other authorities in highly regulated jurisdictions, development of biosimilars has to be accomplished by rigorous and comprehensive comparability exercises in order to assure similarity of the biosimilar with the reference medicinal product in terms of quality characteristics, biological activity, safety and efficacy.
As biologic agents (and thus biosimilars) are produced in living systems (such as bacteria and mammalian cells) using complex manufacturing techniques, biosimilar products are highly similar but not identical to the innovator product.11 This fact seems to be confusing for some physicians and causes concerns with respect to the efficacy and safety of biosimilars.11 This aspect is especially true in IBD, for which no specific clinical trials with the biosimilars have been performed, and regulatory approvals have been granted though extrapolation of data from other indications (rheumatoid arthritis and ankylosing spondylitis).19,20 However, data extrapolation across indications does not imply less reassurance of biosimilar efficacy and safety when supported by scientific justification, but a process for saving time, resources and unnecessary experimental repetition.11 CT-P13 was the first biosimilar to infliximab that obtained regulatory approval by the EMA in 2013 and by the FDA in 2016. It was the first biosimilar approved in the field of rheumatology, dermatology and gastroenterology.
Interchangeability between originator and biosimilarThe legislations for interchangeability and substitution are nonuniform in the USA and the EU. According to the EMA, interchangeability is the medical practice of changing one medicine for another that is expected to achieve the same clinical result in a given clinical setting and in any patient, on the initiative or with the agreement of the prescriber. On the other hand, substitution is the practice of dispensing one medicine instead of another interchangeable medicine at pharmacy level, without consulting the prescriber. In Europe, the EMA indicated that the individual countries in the EU should determine the issue of interchangeability. Thus, this issue is not part of the EMA approval, leaving the decision-making to the discretion of the national authorities. In Europe (and specifically in Spain), automatic substitution of biosimilars is generally not allowed or recommended. However, because the EMA does not have the authority to designate a biosimilar as interchangeable (unlike the FDA, see below), the decisions on interchangeability and/or substitution rely on the competent national authorities.
As defined by the FDA, a biological product is considered to be interchangeable with the reference product if (i) the biological product is biosimilar to the reference product; and (ii) it can be expected to produce the same clinical result in any given patient.16 In addition, for a biological product that is administered more than once to an individual, the risk in terms of safety or diminished efficacy of alternating or switching between the use of the biological product and the reference product is not higher than the risk of using the reference product without such alternation or switch.21 In the US, the federal law has differentiated the approval of products into two stages: (i) the “biosimilar” has to provide evidence of basic similarity to the reference medicinal product, and (ii) an additional approval status called “interchangeable biosimilar” is required to allow for unlimited transition from the reference medicinal product. In other words, biosimilarity does not imply interchangeability, which is much more stringent.22 At present, the FDA has the authority to designate a biosimilar as interchangeable, but substitution is then regulated at state level. According to the FDA, the approval of interchangeability allows that the biosimilar “may be substituted for the reference product without the intervention of the prescribing healthcare provider”.21,23 However, at the present time, the FDA has not yet provided guidance on the method of study needed to enable a determination of interchangeability.
A recent systematic review summarized the evidence about the bioequivalence between biosimilar and reference anti-TNF, and concluded that evidence supports the biosimilarity and interchangeability of biosimilar and reference TNF-inhibitors.24
In the present manuscript, and for practical reasons, we will use the term “switching”, which may be generally defined as a decision by the treating physician to exchange one medicine for another with the same therapeutic intent in patients who are undergoing treatment. We refer to switching as changing a patient on an originator product to a biosimilar one single time, and alternating as taking a patient on an originator product back and forth to a biosimilar multiple times.25
Switching between biologicals different from anti-TNFsSwitching data from a number of randomized and nonrandomized trials consistently show that detrimental effects of switching between reference biologics and their EMA-approved biosimilars –including epoetin,26 granulocyte colony-stimulating factors,27 human growth hormone28 are unlikely to happen. It has been shown that epoetin alfa and epoetin zeta therapy can be interchanged without any clinically significant alteration in efficacy, safety, or epoetin dose, in patients with chronic kidney disease on dialysis receiving stable epoetin maintenance therapy.26 Equivalence of the two filgrastims (originator and biosimilar) was also demonstrated for serum concentration profile, for absolute neutrophil count profile and, even more importantly, for CD34+ cell count, which is a marker for the ability of the granulocyte colony-stimulating factor to mobilize stem cells.27 Finally, patients were successfully switched from originator to biosimilar recombinant human growth hormone (somatropin), with no negative impact on growth, and no serious or unexpected adverse drug reactions.28
Switching between Remicade® and CT-P13 in diseases different from inflammatory bowel diseaseThe infliximab biosimilar CT-P13 was approved based on two clinical trials comparing it with its originator product: the PLANETAS study (in patients with ankylosing spondylitis19), and the PLANETRA study (in patients with rheumatoid arthritis20). Extension studies of the PLANETRA and PLANETAS studies have recently been performed to investigate the long-term efficacy and safety of extended CT-P13 treatment over 2 years, and the efficacy and safety of switching from originator infliximab to CT-P13 for 1 year.29,30
In the PLANETRA extension study,30 the objectives were to assess the efficacy and safety of switching from Remicade® to its biosimilar CT-P13 (Remsima®, Inflectra®) or continuing CT-P13 in patients with rheumatoid arthritis. This open-label extension study recruited patients with rheumatoid arthritis who completed the 54-week, randomized study comparing CT-P13 with Remicade®. Data were analyzed for patients who received CT-P13 for 102 weeks (maintenance group) and for those who received Remicade® for 54 weeks and then switched to CT-P13 (switch group). Response rates at week 102 for maintenance versus switch groups were similar. The proportion of patients with antibodies to infliximab (ATI) was comparable between groups. Treatment-emergent adverse events occurred in similar proportions of patients in the two groups during the extension study. Therefore, the authors concluded that comparable efficacy and tolerability were observed in patients with rheumatoid arthritis who switched from reference product to its biosimilar CT-P13 for an additional year and in those who had long-term CT-P13 treatment for 2 years.
In the PLANETAS extension study,29 with similar aims and design to the PLANETRA extension study, but in patients with ankylosing spondylitis, the response rates at week 102 were comparable between the maintenance and switch groups. Furthermore, ATIs positivity rates were also comparable. Finally, adverse events letting to treatment discontinuation were also similar in both groups. The conclusion of this last study was that switching from Remicade® to its biosimilar CT-P13 is possible without negative effects on safety or efficacy in patients with ankylosing spondylitis.
Other authors have confirmed the encouraging results of this switching strategy in several rheumatic diseases.31 Nikiphorou et al. aimed to gain clinical experience on the switching from Remicade® to CT-P13 in patients with rheumatic disease, and concluded that the clinical effectiveness was comparable, with no safety signals.32 Tanaka et al. also demonstrated, in patients with rheumatoid arthritis, that CT-P13 achieved a stable clinical efficacy and was well tolerated in patients who switched to this drug after 54 weeks of infliximab treatment.33 Moreover, Benucci et al. demonstrated that the switch from innovator to biosimilar infliximab was not associated with any significant difference in efficacy, adverse events or ATI level in patients with spondyloarthritis.34
Finally, it has also been reported that, in patients with psoriasis receiving infliximab originator, treatment can be switched to the infliximab biosimilar without experiencing a significant change in clinical response or additional adverse events.35
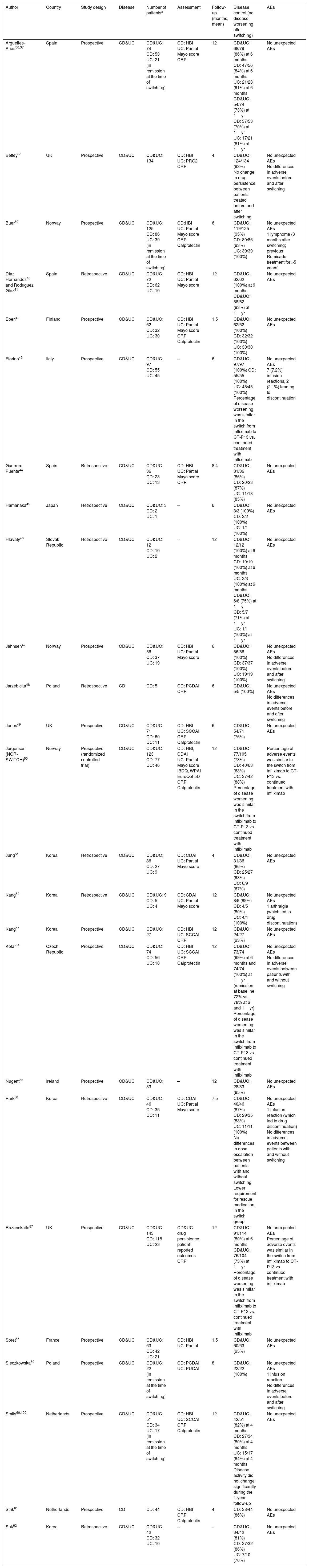
Switching between Remicade® and CT-P13 in inflammatory bowel disease: efficacyThrough electronic and manual bibliographic searches, we identified 24 studies evaluating the switching between anti-TNF originator (Remicade®) and biosimilar (CT-P13) in 1326 patients with IBD.36–62 Seven studies were excluded, as data on the effectiveness in switched patients (to calculate de percentage of “no disease worsening” after switching) were not provided63–68 or infliximab treatment was previously intensified in all patients.69
The characteristics and the results in terms of effectiveness and safety of the included studies evaluating switching in patients with IBD are summarized in Table 2. Regarding the study design, most studies were retrospective, and only one was randomized,50 which will be reviewed in more detail below. Most of the studies included both CD and UC patients. The number of included patients was, in general, quite low, with less than 100 patients in most of the cases; only 4 studies included more than 100 patients, with the largest study including 134 patients for the analysis.38 Effectiveness was evaluated through clinical assessment (mostly by the Harvey–Bradshaw index in CD and the Partial Mayo score in UC), with no endoscopic evaluation generally available. In some cases, changes in C-reactive protein and fecal calprotectin were also analyzed. Follow-up ranged from 1.5 to 12 months, being between 6 and 12 months in most of the studies.
Studies evaluating the switching between anti-TNF originator (Remicade®) and biosimilar (CT-P13).
| Author | Country | Study design | Disease | Number of patientsa | Assessment | Follow-up (months, mean) | Disease control (no disease worsening after switching) | AEs |
|---|---|---|---|---|---|---|---|---|
| Arguelles-Arias36,37 | Spain | Prospective | CD&UC | CD&UC: 74 CD: 53 UC: 21 (in remission at the time of switching) | CD: HBI UC: Partial Mayo score CRP | 12 | CD&UC: 68/79 (86%) at 6 months CD: 47/56 (84%) at 6 months UC: 21/23 (91%) at 6 months CD&UC: 54/74 (73%) at 1yr CD: 37/53 (70%) at 1yr UC: 17/21 (81%) at 1yr | No unexpected AEs |
| Bettey38 | UK | Prospective | CD&UC | CD&UC: 134 | CD: HBI UC: PRO2 CRP | 4 | CD&UC: 124/134 (93%) No change in drug persistence between patients treated before and after switching | No unexpected AEs No differences in adverse events before and after switching |
| Buer39 | Norway | Prospective | CD&UC | CD&UC: 125 CD: 86 UC: 39 (in remission at the time of switching) | CD:HBI UC: Partial Mayo score CRP Calprotectin | 6 | CD&UC: 119/125 (95%) CD: 80/86 (93%) UC: 39/39 (100%) | No unexpected AEs 1 lymphoma (3 months after switching; previous Remicade treatment for >5 years) |
| Díaz Hernández40 and Rodríguez Glez41 | Spain | Retrospective | CD&UC | CD&UC: 72 CD: 62 UC: 10 | CD: HBI UC: Partial Mayo score | 12 | CD&UC: 62/62 (100%) at 6 months CD&UC: 58/62 (93%) at 1yr | No unexpected AEs |
| Eberl42 | Finland | Prospective | CD&UC | CD&UC: 62 CD: 32 UC: 30 | CD: HBI UC: Partial Mayo score CRP Calprotectin | 1.5 | CD&UC: 62/62 (100%) CD: 32/32 (100%) UC: 30/30 (100%) | No unexpected AEs |
| Fiorino43 | Italy | Prospective | CD&UC | CD&UC: 97 CD: 55 UC: 45 | – | 6 | CD&UC: 97/97 (100%) CD: 55/55 (100%) UC: 45/45 (100%) Percentage of disease worsening was similar in the switch from infliximab to CT-P13 vs. continued treatment with infliximab | No unexpected AEs 7 (7.2%) infusion reactions, 2 (2.1%) leading to discontinuation |
| Guerrero Puente44 | Spain | Retrospective | CD&UC | CD&UC: 36 CD: 23 UC: 13 | CD: HBI UC: Partial Mayo score CRP | 8.4 | CD&UC: 31/36 (86%) CD: 20/23 (87%) UC: 11/13 (85%) | No unexpected AEs |
| Hamanaka45 | Japan | Retrospective | CD&UC | CD&UC: 3 CD: 2 UC: 1 | – | 6 | CD&UC: 3/3 (100%) CD: 2/2 (100%) UC: 1/1 (100%) | No unexpected AEs |
| Hlavatý46 | Slovak Republic | Retrospective | CD&UC | CD&UC: 12 CD: 10 UC: 2 | – | 12 | CD&UC: 12/12 (100%) at 6 months CD: 10/10 (100%) at 6 months UC: 2/3 (100%) at 6 months CD&UC: 6/8 (75%) at 1yr CD: 5/7 (71%) at 1yr UC: 1/1 (100%) at 1yr | No unexpected AEs |
| Jahnsen47 | Norway | Prospective | CD&UC | CD&UC: 56 CD: 37 UC: 19 | CD: HBI UC: Partial Mayo score | 6 | CD&UC: 56/56 (100%) CD: 37/37 (100%) UC: 19/19 (100%) | No unexpected AEs No differences in adverse events before and after switching |
| Jarzebicka48 | Poland | Retrospective | CD | CD: 5 | CD: PCDAI CRP | 6 | CD&UC: 5/5 (100%) | No unexpected AEs No differences in adverse events before and after switching |
| Jones49 | UK | Prospective | CD&UC | CD&UC: 71 CD: 60 UC: 11 | CD: HBI UC: SCCAI CRP Calprotectin | 6 | CD&UC: 54/71 (76%) | No unexpected AEs |
| Jorgensen (NOR-SWITCH)50 | Norway | Prospective (randomized controlled trial) | CD&UC | CD&UC: 123 CD: 77 UC: 46 | CD: HBI, CDAI UC: Partial Mayo score IBDQ, WPAI EuroQol-5D CRP Calprotectin | 12 | CD&UC: 77/105 (73%) CD: 40/63 (63%) UC: 37/42 (88%) Percentage of disease worsening was similar in the switch from infliximab to CT-P13 vs. continued treatment with infliximab | Percentage of adverse events was similar in the switch from infliximab to CT-P13 vs. continued treatment with infliximab |
| Jung51 | Korea | Retrospective | CD&UC | CD&UC: 36 CD: 27 UC: 9 | CD: CDAI UC: Partial Mayo score | 4 | CD&UC: 31/36 (86%) CD: 25/27 (93%) UC: 6/9 (67%) | No unexpected AEs |
| Kang52 | Korea | Retrospective | CD&UC | CD&UC: 9 CD: 5 UC: 4 | CD: CDAI UC: Partial Mayo score | 12 | CD&UC: 8/9 (89%) CD: 4/5 (80%) UC: 4/4 (100%) | No unexpected AEs 1 arthralgia (which led to drug discontinuation) |
| Kang53 | Korea | Prospective | CD&UC | CD&UC: 27 | CD: HBI UC: SCCAI CRP | 12 | CD&UC: 24/27 (93%) | No unexpected AEs |
| Kolar54 | Czech Republic | Prospective | CD&UC | CD&UC: 74 CD: 56 UC: 18 | CD: HBI UC: SCCAI CRP Calprotectin | 12 | CD&UC: 73/74 (99%) at 6 months and 74/74 (100%) at 1yr (remission at baseline 72% vs. 78% at 6 and 1yr) Percentage of disease worsening was similar in the switch from infliximab to CT-P13 vs. continued treatment with infliximab | No unexpected AEs No differences in adverse events between patients with and without switching |
| Nugent55 | Ireland | Prospective | CD&UC | CD&UC: 33 | – | 12 | CD&UC: 28/33 (85%) | No unexpected AEs |
| Park56 | Korea | Retrospective | CD&UC | CD&UC: 46 CD: 35 UC: 11 | CD: CDAI UC: Partial Mayo score | 7.5 | CD&UC: 40/46 (87%) CD: 29/35 (83%) UC: 11/11 (100%) No differences in dose escalation between patients with and without switching Lower requirement for rescue medication in the switch group | No unexpected AEs 1 infusion reaction (which led to drug discontinuation) No differences in adverse events between patients with and without switching |
| Razanskaite57 | UK | Prospective | CD&UC | CD&UC: 143 CD: 118 UC: 23 | CD&UC: drug persistence; patient reported outcomes CRP | 12 | CD&UC: 91/114 (80%) at 6 months CD&UC: 76/104 (73%) at 1yr Percentage of disease worsening was similar in the switch from infliximab to CT-P13 vs. continued treatment with infliximab | No unexpected AEs Percentage of adverse events was similar in the switch from infliximab to CT-P13 vs. continued treatment with infliximab |
| Soret58 | France | Prospective | CD&UC | CD&UC: 63 CD: 42 UC: 21 | CD: HBI UC: Partial | 1.5 | CD&UC: 60/63 (95%) | No unexpected AEs |
| Sieczkowska59 | Poland | Prospective | CD&UC | CD&UC: 22 (in remission at the time of switching) | CD: PCDAI UC: PUCAI | 8 | CD&UC: 22/22 (100%) | No unexpected AEs 1 infusion reaction No differences in adverse events before and after switching |
| Smits60,100 | Netherlands | Prospective | CD&UC | CD&UC: 51 CD: 34 UC: 17 (in remission at the time of switching) | CD: HBI UC: SCCAI CRP Calprotectin | 12 | CD&UC: 42/51 (82%) at 4 months CD: 27/34 (80%) at 4 months UC: 15/17 (84%) at 4 months Disease activity did not change significantly during the 1-year follow-up | No unexpected AEs |
| Strik61 | Netherlands | Prospective | CD | CD: 44 | CD: HBI CRP Calprotectin | 4 | CD: 38/44 (86%) | No unexpected AEs |
| Suk62 | Korea | Retrospective | CD&UC | CD&UC: 42 CD: 32 UC: 10 | – | – | CD&UC: 34/42 (81%) CD: 27/32 (86%) UC: 7/10 (70%) | No unexpected AEs |
Number of patients with follow-up available to calculate the endpoint of efficacy and/or safety.
AEs: adverse events; CD: Crohn's disease; UC: ulcerative colitis; CDAI: Crohn's disease activity index; HBI: Harvey–Bradshaw index; PCDAI: Pediatric Crohn's disease activity index; SCCAI: simple clinical colitis activity index; CRP: C-reactive protein; PRO: patient reported outcomes; IBDQ: Inflammatory Bowel Diseases Questionnaire; WPAI: Work Productivity and Activity Impairment.
From the 1326 patients available to calculate effectiveness (Table 2), disease control (that is, no disease worsening after switching) was confirmed in 1163 patients (weighted mean, 88%; 95% confidence interval, from 86% to 89%). When only studies including a more homogeneous follow-up from 4 to 8 months were included, this figure was 90% (89–92%). When a subanalysis was conducted only for CD patients, the proportion of patients maintaining disease control was 86% (82–89%), and the corresponding figure for UC patients was 93% (89–96%).
A previous systematic review aimed to analyze the data from available clinical trials that investigated the validity of indication extrapolation of CT-P13 for the treatment of IBD in naïve patients and in patients who switched from its originator infliximab.70 However, the literature search was performed only until September 2016, and therefore, at that time, the number of studies including only IBD patients switching between anti-TNF originator and biosimilar was very low (only three).70
Another systematic review evaluated the efficacy of biosimilars in both anti-TNF naïve and in Remicade® exposed (switched) IBD patients.71 The pooled rates of sustained clinical response among CD and UC after switching from infliximab to CT-P13 at 30–32 weeks were 85% and 96%, respectively; and at 48–63 weeks were 75% and 83%. Percentages for sustained clinical remission rates at 16 and 51 weeks were, for CD, 74% and 92%; and for UC, 62% and 83%. All these figures appear to be comparable to the rates seen with infliximab originator. However, again, as the literature search for this last meta-analysis was performed only until May 2016, the number of studies was very low (overall, only six studies switching from infliximab originator to biosimilar, meta-analysis being based at most on three studies), and the corresponding number of included patients was also quite small (N=277). In our present review, 24 studies including 1326 patients have been included.
Switching between Remicade® and CT-P13 in inflammatory bowel disease: safety and immunogenicityNo unexpected adverse events were reported in any of the included studies assessing the switching strategy in IBD patients.36–62 When evaluated, no differences in adverse events before and after switching were reported either. Furthermore, no differences in adverse events between patients with and without switching were confirmed (in those studies including these two subpopulations). Finally, in the only randomized controlled trial study performed up to now (the NOR-SWITCH trial), the frequencies of reported adverse events were not different between patients with and without switching.50
In addition to effectiveness, the other main concern of switching patients from a reference product to its biosimilar is immunogenicity. A recent systematic review reported no increase in ATIs formation in rheumatoid arthritis patients treated with biosimilars and concluded that immunogenicity seemed comparable across treatment groups in all studies.24 It concluded that despite the paucity of studies, the existing evidence supports the biosimilarity and interchangeability of these newly developed TNF-inhibitor products, especially for the treatment of patients with rheumatoid arthritis.
Ben-Horin et al. studied the cross-reactivity of antibodies to Remicade® and CT-P13 in patients with IBD.72 Serum samples obtained from patients with IBD and healthy individuals (negative controls) were tested. The results showed that anti-Remicade® antibodies recognize and functionally inhibit CT-P13 to a similar degree than the originator, suggesting similar immunogenicity, and shared immunodominant epitopes on these two infliximab agents. Finally, a study by Gils et al. concluded that the assay for measuring Remicade® can also be used to determine Remsima® and Inflectra® concentrations, and that in all patients with IBD who develop anti-Remicade® antibodies, the antibodies cross-react with infliximab biosimilars.73 Jung et al. confirmed that all patients who had detectable ATIs at baseline (measured prior the first infusion of CT-P13) had detectable ATIs against CT-P13 during follow-up, which may suggest cross-reactivity of ATIs.60 These were expected findings, as Remicade® and CT-P13 share an identical amino acid sequence and a highly similar three-dimensional structure.74 Other authors have confirmed that originator and biosimilar infliximab have comparable immunogenicity in patients with IBD.75 Therefore, patients with IBD who develop high-titer of ATIs and infusion reaction/loss of response to Remicade® should not be considered for switching to CT-P13.
Finally, no higher immunogenicity after switching has been reported in several studies.39,42,44,50,53,54,57,58,76,77 In particular, the incidence of ATIs detected during the NOR-SWITCH trial was 7.1% and 7.9% in the originator and CT-P13 patients, respectively.50 Accordingly, several authors have reported no change in infliximab serum level (and the frequency of adverse events, including infusion reactions) detected after switching.39,42,44,50,53,54,57–59,61
No apparent new safety or immunogenicity signals or changes in efficacy arised after a single switch. This is unlike multiple repeated transitions between a reference product and its biosimilar, which should be discouraged because of absence of data on its safety and because of the significant challenges to agent-specific surveillance when multiple transitions are performed.10
Because repeated switches may increase the likelihood of developing ATIs, which in turn can lead to compromised safety and efficacy, trial designs incorporating multiple switches between originator and biosimilar products would provide additional and clinically relevant information about the immunogenicity of biologics.25
Methodological limitations of switching studies in inflammatory bowel diseaseThe aforementioned studies evaluating the switching between anti-TNF originator (Remicade®) and biosimilar (CT-P13) in patients with IBD should be interpreted with caution, as they have several relevant limitations, among them:
- 1)
The first and obvious limitation is the low sample size of the individual studies. As previously mentioned, the number of patients included in each study was quite low (less than 100 patients in most of the cases), with the largest study including only 134 valid patients. Therefore, larger patient numbers are necessary for confirming this clinical experience.
- 2)
Secondly, the follow-up duration of the studies was relatively short. Thus, follow-up was generally of only 6–12 months, and the maximum follow-up was of 12 months (in 10 studies). Low incidence adverse drug reactions may require large numbers of patients followed for years to determine risk. Therefore, it is obvious that longer follow-up is required to assess not only efficacy, but also safety.
- 3)
The heterogeneous time of switching from Remicade® to CT-P13 during the course of therapy – both among different studies and also within individual patients in each study – may further limit the conclusions of the studies.
- 4)
Effectiveness was evaluated through clinical assessment, or sometimes with additional biological parameters (such as C-reactive protein and fecal calprotectin), but more objective – endoscopic – evaluation was not generally performed.
- 5)
A main drawback is that most of the studies were retrospective. Furthermore, excepting the NOR-SWITCH trial, the design of the studies did not include a true control group that allowed patients to continue Remicade®. Thus, to assess effectiveness, the available studies calculated the percentage of patients with “disease control” (that is, with “no disease worsening” after switching) during follow-up. It is true that some studies reported “no change in drug persistence between patients treated before and after switching” or found “no differences in dose escalation between patients with and without switching”, but these were not true (randomized) controlled studies. Therefore, it is difficult to interpret changes in efficacy (and safety, and pharmacokinetics) that either may be due to the switch to CT-P13 or may be coincident with the natural course of the disease.78
- 6)
Excepting the NOR-SWITCH trial, all studies were unblinded. Subjective reasons (negative expectations) may play a role in disease worsening and among discontinuations of biosimilars. Thus, in clinical practice, non-medical switching of biological medication may provoke nocebo effects due to unexplained deterioration of therapeutic benefits.79
- 7)
Finally, no studies have addressed so far efficacy, safety, and immunogenicity of cross-switching (switching between two biosimilars), reverse-switching (switching from a biosimilar to its originator), or multiples or repeated switches.
The NOR-SWITCH trial is, up to now, the only randomized controlled trial that has compared, in IBD patients, Remicade® and CT-P13.50 This is an independent study sponsored by the Norwegian government. Patients with CD, UC, rheumatoid arthritis, spondyloarthritis, psoriatic arthritis, and chronic plaque psoriasis on stable treatment with the originator infliximab (Remicade®) for at least 6 months were eligible. It was designed as a non-inferiority trial with a non-inferiority margin set to 15%. Power calculations indicated that 394 patients were required in the primary per-protocol set. Patients with informed consent were randomized 1:1 to either continue originator infliximab or switch to CT-P13 treatment using an unchanged dosing regimen. The primary endpoint was disease worsening during follow-up (52 weeks) according to a worsening in disease-specific composite measures and/or a consensus between investigator and patient leading to major change in treatment. Exploratory subgroup analyses were performed to examine disease worsening within each of the six diagnoses. Disease worsening in CD was defined as an increase in Harvey–Bradshaw index of ≥4 points from randomization and a minimum score of 7 points, and in UC as an increase in partial Mayo score of ≥3 points from randomization and a minimum score of ≥5 points. If a patient did not fulfill the formal definition, but experienced a clinically significant worsening according to both the investigator and patient, which led to a major change in treatment, this was considered as a disease worsening. Among the secondary endpoints, erythrocyte sedimentation rate, C-reactive protein, and calprotectin were determined; in addition, remission status and disease activity according to CDAI were also considered. Finally, the Inflammatory Bowel Diseases Questionnaire (IBDQ), the quality of life (EQ-5D) and the work productivity (WPAI) were also calculated.
This study enrolled 481 patients, from 40 Norwegian centers. At 52 weeks, disease worsening occurred in 26.2% and 29.6% of patients in the originator and CTP13 arms, respectively (difference: 4.4%, 95% CI from −12.7 to 3.9%). The frequency of disease worsening in each specific diagnosis, and changes in the generic disease variables and disease-specific composite measures, were not different in either of the arms. In particular, 155 patients with CD and 93 patients with UC were included. Disease worsening was reported in 21.2% of CD patients treated with Remicade® and in 36.5% of those treated with biosimilar (difference: −14.3%, 95% CI from −29.3 to 0.7%). On the other hand, disease worsening was reported in 9.1% of UC patients treated with Remicade® and in 11.9% of those treated with biosimilar (difference: 2.8%, 95% CI from −15.2 to 10.0%). The authors concluded that switch from Remicade® to CT-P13 was not inferior to continued treatment with Remicade®. An open-label 6-month follow-up of this study, where patients having had Remicade® for one year were switched to infliximab biosimilar, is ongoing.
Although the NOR-SWITCH trial is perceived by some as a critical study in the determination of whether it is effective and safe to switch biologic therapy (mainly because it is the only randomized controlled trial in IBD), it has, however, significant design limitations25; the most relevant can be summarized as follows:
- 1)
Type of disease: The diversity of patients included in the NOR-SWITCH trial, including patients with rheumatoid arthritis, spondyloarthritis, psoriatic arthritis, chronic plaque psoriasis, UC and CD may drive potential bias. It has been questioned whether this approach could limit the ability to draw firm conclusions as to the effects of the switch, since each illness is very different with regard to duration of response and clinical measures. In this respect, all NOR-SWITCH patients were required to be stable on current infliximab therapy for 6 months, but the protocol (ClinicalTrials.gov Identifier: NCT02148640) fails to consider that criteria for stable patients require definition and are typically disease-specific.25
- 2)
Baseline characteristics regarding disease activity and medication dose: Although it is stated in the protocol that patients were on stable treatment with the originator infliximab for at least 6 months, not all patients were in clinical remission at that moment (at baseline, only approximately 65% of patients with CD, and 90% of those with UC, were in clinical remission). In this same respect, we do not know either whether the infliximab dose could have been previously optimized (changes in dose or frequency of infliximab) at any time before switching occurs, and, in that case, whether the proportion of this dose intensification was equally distributed between the two groups (switchers and non-switchers).
- 3)
Co-treatment with immunosuppressive drugs: The proportion of patients (overall, considering all included diseases) receiving concomitant immunosuppressive medication was slightly higher in the biosimilar group than in the originator one (54% vs. 47%, 7% difference), and this may bias results toward better results in the biosimilar group. This tendency was also true for CD (47% vs. 38% for patients in the biosimilar and the originator, respectively).
- 4)
Outcome measures: As it has been accurately stated by Faccin et al.,25 assessments of the primary outcome (disease worsening, measured with variety of instruments depending on the specific disease) are not universally accepted clinical definitions. “Major” changes in concomitant medications are to be considered as indicators of disease worsening, but “major” is poorly defined. Furthermore, at the analysis stage, grouping results from the aforementioned six diseases may create bias, complicating the interpretation of results because no end point is common to all diseases examined. It has even been suggested (see “The design of clinical trials to support the switching and alternation of biosimilars” by Faccin et al.25) that achieving the overall comparability objective cannot be directly translated to any of the individual indications, as a result of potentially insufficient sample sizes in each indication, and that therefore, the non-inferiority margin is not interpretable to the individual indication (see next point).
- 5)
Statistical limitations: Most of the statistical concerns deal with the calculation of the sample size, which was based on an estimated rate of disease worsening of 30%, a number not observed in previous literature, at least in IBD.78,80 The incidence rate of loss of response to infliximab has been estimated to be of approximately 13% per patient-year of follow-up.78 This rate seems to be higher during the first year, but progressively decreases with time.78,80 As patients being potential candidates for switching are those that have been in clinical remission for at least some months (in the NOR-SWITCH, patients had to be on stable treatment with Remicade® for at least 6 months), they might have a lower incidence rate of loss of response, which could be estimated to be of only approximately 10%.80 Therefore, individual estimates for each anti-TNF indication – for each disease – would have been more appropriate; in other words, different sample sizes should have been required based on indication-specific non-inferiority margins, as it is unlikely that patient populations for all indications are homogenous.25 Furthermore, the study was designed as a non-inferiority trial with a non-inferiority margin set to 15%; however, it could be argued that this selected margin (15%) is insufficiently stringent, and that a 10% difference may be considered clinically significant (indicating that the power calculations should have included a higher number of patients). In this respect, disease worsening (the primary endpoint in the NOR-SWITCH trial) in CD patients was reported in 21.2% of patients treated with Remicade® and in 36.5% of those treated with biosimilar (risk difference: −14.3%, 95% CI from −29.3 to 0.7%). Finally, losses to follow-up, which happened in approximately 15% of cases, may further aggravate the problem of the possible insufficient sample size. In this respect, intention-to-treat analyses, in addition to per-protocol ones, should always be provided. Although per-protocol analyses tend to enhance treatment differences and are usually used for non-inferiority or equivalence trials, sensitivity analyses with an intent-to-treat population should show consistent outcomes.25 Fortunately, secondary efficacy analyses were performed by the authors in the full analysis set, consisting of eligible randomized patients who received at least one infusion after randomization, and the results were equivalent in terms of the primary outcome (disease worsening): 17.9% of patients treated with Remicade® and in 31.2% of those treated with biosimilar (risk difference: −12.7%, 95% CI from −25.8 to 0.5%).50
In summary, the NOR-SWITCH trial concluded that switch from Remicade® to CT-P13 was not inferior to continued treatment with Remicade®. However, because of the design limitations of this trial, data must be interpreted with caution. In particular, it should be taken into account that addressing multiple indications in one trial may be a flaw that could prevent drawing a meaningful conclusion of the effect of switching for any of the indicated diseases, such as CD or UC. Therefore, more robust data would be desirable to provide clinically meaningful information about the safety and efficacy of switching between originator and biosimilar therapies in IBD.
Switching in inflammatory bowel disease: ongoing studiesFortunately, several ongoing studies will soon provide additional information on the efficacy and safety of switching in patients with IBD; among them, the following stand out:
“A Randomized, Double-Blind, Parallel-Group, Phase 3 Study to Demonstrate Noninferiority in Efficacy and to Assess Safety of CT-P13 Compared to Remicade in Patients With Active Crohn's Disease” (ClinicalTrials.gov Identifier: NCT02096861). The aim of this study is to assess non-inferiority in efficacy and to assess overall safety of CT-P13 compared to Remicade® in patients with active CD up to Week 54. In this study, a randomized transition from originator infliximab to CT-P13 as well as from CT-P13 to originator infliximab will be evaluated.
“Evaluation of the Switch From the Original Infliximab (Remicade®) to Its Biosimilar (Inflectra®) in Daily Practice at Cochin Hospital” (ClinicalTrials.gov Identifier: NCT02998398). The purpose of this study is to evaluate the effectiveness of the switch from Remicade® to Inflectra® in all the patients at Cochin hospital receiving Remicade® for rheumatic, gastro-enterologic or ophthalmic condition.
“Efficacy and Safety of Infliximab-biosimilar (Inflectra®) Compared to Infliximab-innovator (Remicade®) in Patients With Inflammatory Bowel Disease in Remission: the SIMILAR Trial” (ClinicalTrials.gov Identifier: NCT02452151). The objective of this study is to compare the efficacy of infliximab-biosimilar to infliximab-innovator and to demonstrate its non-inferiority, in patients with CD or UC in remission under treatment with infliximab up to 3 months.
Cost saving of switching in inflammatory bowel diseaseThe reduced price of biosimilars can lead to cost efficiencies and drive competition. In turn, this may benefit healthcare systems and improve patient care by increasing access to biologic therapy. With growing numbers of biosimilar products there are now more options for healthcare providers and patients, not only to access biological products earlier, but also to possibly switch from costly originator versions to biosimilar alternatives. Theoretically, biosimilars of monoclonal antibodies could reduce the price per mg from 25% to 40%, a considerable difference, adding “value” to the treatment.81 However, market forces are complex because regulators, payers, providers, physicians, pharmacists, and patients have different roles and input in different geographical areas.81
Some studies have demonstrated that, specifically in patients with IBD, the switch from the infliximab originator to the biosimilar can yield substantial cost savings.38,40,82,83 In this respect, the aim of a recent study was to evaluate the one-year budget impact of introducing biosimilar infliximab in the management of CD and UC from the healthcare system perspective.83 An Excel-based budget impact model with a one-year time horizon was developed. The numbers of patients eligible for infliximab were calculated based on disease incidence and prevalence rates in Germany, Italy, Belgium, the Netherlands and the UK. For the price of biosimilar infliximab, 3 discount scenarios versus infliximab (10%, 20%, and 30%) were applied. For patients currently treated with infliximab (switch population), the biosimilar infliximab market share was assumed to be 25% in all scenarios. For the switch population, the corresponding projected savings ranged from € 0.7 million (Italy) to € 16.1 million (Germany) for CD, and from € 0.3 million (UK) to € 5.3 million (Germany) for UC. Therefore, the authors concluded that the introduction of biosimilar infliximab as a treatment option for patients with IBD could achieve substantial cost savings for healthcare systems.
Recommendations of health and scientific organizations on switching in inflammatory bowel diseaseSome scientific and health organizations have positioned against switching in IBD (although in most of the cases these recommendations were published several years ago, which may partially explain this criterion), including: the Canadian Association of Gastroenterology (year 2013),84 the Italian Group of Inflammatory Bowel Disease (year 2014),85 the Spanish Society of Digestive Diseases (year 2013),86 the Polish National Consultant in Gastroenterology (year 2014),87 and the European Society for Paediatric Gastroenterology Hepatology and Nutrition European Paediatric IBD Porto Group (year 2015).88 The French Medicines Agency89 allowed it when initiating a course of treatment and only if the prescribing physician has not marked the prescription as “non-substitutable”.
However, more recent statements tend to support and recommend switching in IBD, such as the following: the European Crohn's and Colitis Organisation (year 2017),90 the British Society of Gastroenterology (year 2016; approved the switching of stable patients on reference infliximab to the biosimilar, but recommended against automatic substitution by a pharmacy without the involvement of the prescribing physician),91 the Finnish Medicines Agency (FIMEA) (year 2015; allowed under supervision of a health care professional),92 the Dutch Medicines Agency (year 2015; allowed under supervision of a health care professional),93 the Australian Medicines Agency (year 2015),94 the Italian Medicines Agency (2012; allowed under supervision of a health care professional),95 the Portuguese Association of Hospital Pharmacists (2016; if a drug is approved as a biosimilar, this decision should be interpreted as the result of an extensive comparability exercise establishing a therapeutic equivalence to the original drug, and from a regulatory point of view, this decision means that these drugs are interchangeable).96
In particular, the European Crohn's and Colitis Organisation, in the “ECCO Position Statement on the Use of Biosimilars for Inflammatory Bowel Disease-An Update”, which has just been published in 2017,90 states that: “Switching from the originator to a biosimilar in patients with IBD is acceptable“; and that “Switching from originator to a biosimilar should be performed following appropriate discussion between physicians, nurses, pharmacists, and patients, and according to national recommendation”.
The opinion of the patientLast but not least, the opinion of the patient should always be taken into consideration. During 2015, the Gastrointestinal Society, a patient group from Canada, hosted a survey on the use of biologics/biosimilars for IBD.97 Questions included their understanding and opinions regarding subsequent entry biologics, including the possibility of switching from an innovator biologic to a biosimilar. Only 52% of patients believed that having the same international non-proprietary name implied that patients could safely switch between the products during a course of treatment and expect the same effectiveness and safety. Moreover, 95% of those surveyed said that it is important their physician, together with them, have the sole authority to decide the most suitable biologic medication to treat their disease.97 In this respect, a recent study indicates that, in most cases, the patient feels confident with his/her doctor's recommendation.68 In this respect, very recently, Bettey et al. evaluated the outcomes of a managed switching program changing IBD patients established on originator infliximab to biosimilar infliximab.38 The most remarkable finding was that all 134 patients agreed to the switch. After switching, one patient requested to change back to the originator infliximab because of flu-like symptoms, and one because of deranged liver function tests.
Physicians must take an active role in educating their patients concerning policies affecting access to biologics and biosimilars. The decision to initiate a biologic, biosimilar, or biosimilar switch, should always take into account patient preference.98 The information offered must be transparent and the requirement of a non-medical switch must be made clear to the patient e.g. financial savings or additional services attached to the switch.90
ConclusionsBiosimilar development has been set out to accomplish two main goals: reduce the expenditure of healthcare on costly biological treatments and improve patient access to these drugs. Thus, the availability of biosimilars represents an unprecedented opportunity to reduce healthcare costs and expand treatment options. In turn, the cost reduction represented by biosimilars promotes industry competition and improves treatment access with sustained quality of care.
Current evidence from real-world IBD cohorts suggests that effectiveness and safety is similar between the infliximab biosimilar and the reference medicinal product. On the other hand, limited data are available concerning Remicade®-treated IBD patients who switched to CT-P13. Observational studies, registries, cohorts and real-world experiences evaluating safety and efficacy upon switching to CT-P13 (and to other biosimilars) showed that there are no concerns relating to safety or efficacy in patients with different types of immune-mediated diseases.99 Specifically in IBD, there have been no reports that switching patients from the reference to the biosimilar infliximab has led to problems, such as increased immunogenicity or any other safety concerns.
Most of this switch data is derived from observational cohorts and open-label studies. The NOR-SWITCH trial is, up to now, the first and only randomized controlled trial assessing this topic, and has concluded that switch from Remicade® to CT-P13 was not inferior to continued treatment with Remicade®. However, it should be taken into account that the NOR-SWITCH was not powered to provide definite conclusions about the effects of the switch within each specific disease, such as IBD. Furthermore, experience on the switching from Remicade® to other infliximab biosimilar different from CT-P13 is lacking, as it is also unknown whether the results of the switching will be similar with other anti-TNFs such as adalimumab.
In conclusion, the risks of switching to biosimilar products seem to be theoretical and not supported by the limited real world safety experience so far. On the contrary, an increasing number of publications have shown that there are no safety or efficacy concerns about switching. Therefore, we think, in accordance with the European Crohn's and Colitis Organisation Position Statement, that switching from the originator to a biosimilar in patients with IBD may be considered “acceptable”. Anyhow, switch should only be considered after the disease is well controlled with Remicade® during a sufficient time.
Finally, decisions about switching should remain in the hands of the treating physician. Moreover, switching between products should be carried out under supervision and monitoring as is done with initiation of any biological drug. Obviously, more robust data, from well designed studies, are required to better assess the efficacy and safety of switching therapies. Furthermore, postmarketing surveillance and registry studies monitoring the safety of biosimilar in patients with IBD who have switched from reference product will be necessary.
Conflict of interestJP Gisbert has served as a speaker, a consultant and advisory member for or has received research funding from MSD, Abbvie, Hospira, Pfizer, Kern Pharma, Biogen, Takeda, Janssen, Roche, Celgene, Ferring, Faes Farma, Shire Pharmaceuticals, Dr. Falk Pharma, Tillotts Pharma, Chiesi, Casen Fleet, Gebro Pharma, Otsuka Pharmaceutical, Vifor Pharma.
M Chaparro has served as a speaker, or has received research or education funding from MSD, Abbvie, Hospira, Pfizer, Takeda, Janssen, Ferring, Shire Pharmaceuticals, Dr. Falk Pharma, Tillotts Pharma.
