




El tumor fibroso solitario (antiguo hemangiopericitoma) es una infrecuente neoplasia de partes blandas cuya localización más frecuente es el retroperitoneo y las extremidades inferiores. Presentamos dos casos de hemangiopericitomas retroperitoneales diagnosticados tras intervención quirúrgica. Realizamos una revisión bibliográfica exponiendo los aspectos clínicos, anatomopatológicos y terapéuticos de estos tumores.
Solitary fibrous tumor (formerly known as hemangiopericytoma) is a rare soft tissue neoplasm, most frequently arising from the retroperitoneum and lower extremities. We present two cases of retroperitoneal solitary fibrous tumors diagnosed after surgical removal. We provide a literature review showing the basic clinical, pathologic and therapeutic features of these tumors.
El tumor fibroso solitario (antiguo hemangiopericitoma) es una rara neoplasia de partes blandas. Puede originarse en cualquier punto del organismo aunque sus localizaciones más frecuentes son el retroperitoneo y las extremidades inferiores. Generalmente se presenta como una masa bien definida, sólida e hipervascularizada1–3.
Observación clínicaCaso 1Mujer de 71 años diagnosticada de colitis ulcerosa hace 40 años, desde entonces asintomática, acude a consultas de digestivo por dolor abdominal de un mes de evolución, más acusado en flanco derecho, astenia, anorexia, pérdida de peso y fiebre vespertina. Se realiza endoscopia oral, colonoscopia y tomografía computarizada (TC) abdominal con contraste intravenoso. La endoscopia oral tan solo revela hernia de hiato por deslizamiento y atrofia difusa de la mucosa gástrica, habiéndose explorado hasta la rodilla inferior. En la colonoscopia se visualizan alteraciones sugestivas de colitis ulcerosa crónica inactiva. La TC abdominal pone de manifiesto una masa de 9×8,5×8cm, heterogénea, con áreas hipodensas sugestivas de necrosis. La masa desplazaba la cabeza del páncreas hacia la izquierda y a las venas cava y renal derecha posteriormente; contactaba con la porción más inferior del lóbulo hepático derecho, aunque no parecía depender del mismo, siendo más probable su origen en la segunda porción duodenal (fig. 1). Tras tumorectomía y escisión de la segunda porción duodenal, la anatomía patológica informó de tumor fibroso solitario retroperitoneal con patrón vascular hemangiopericitoide. La tumoración estaba bien delimitada, no encapsulada, y fue descrita como una proliferación de células fusiformes ordenadas de manera irregular con focos de colagenización y necrosis, presentando áreas hipercelulares con atipia, pleomorfismo celular y aumento del índice mitótico separadas de otras zonas hipocelulares colagenizadas y con proliferación vascular con patrón de crecimiento tipo hemangiopericitoma en la periferia. La inmunohistoquímica presentó positividad para CD34, CD99, Bcl-2 y vimentina. Se demostró positividad en la pared vascular para actina de músculo liso y positividad nuclear de ki-67 en el 40–60% de las células neoplásicas, siendo indicativo de una tasa de proliferación media. Fueron negativos CD68, C-Kit, S-100 y desmina.
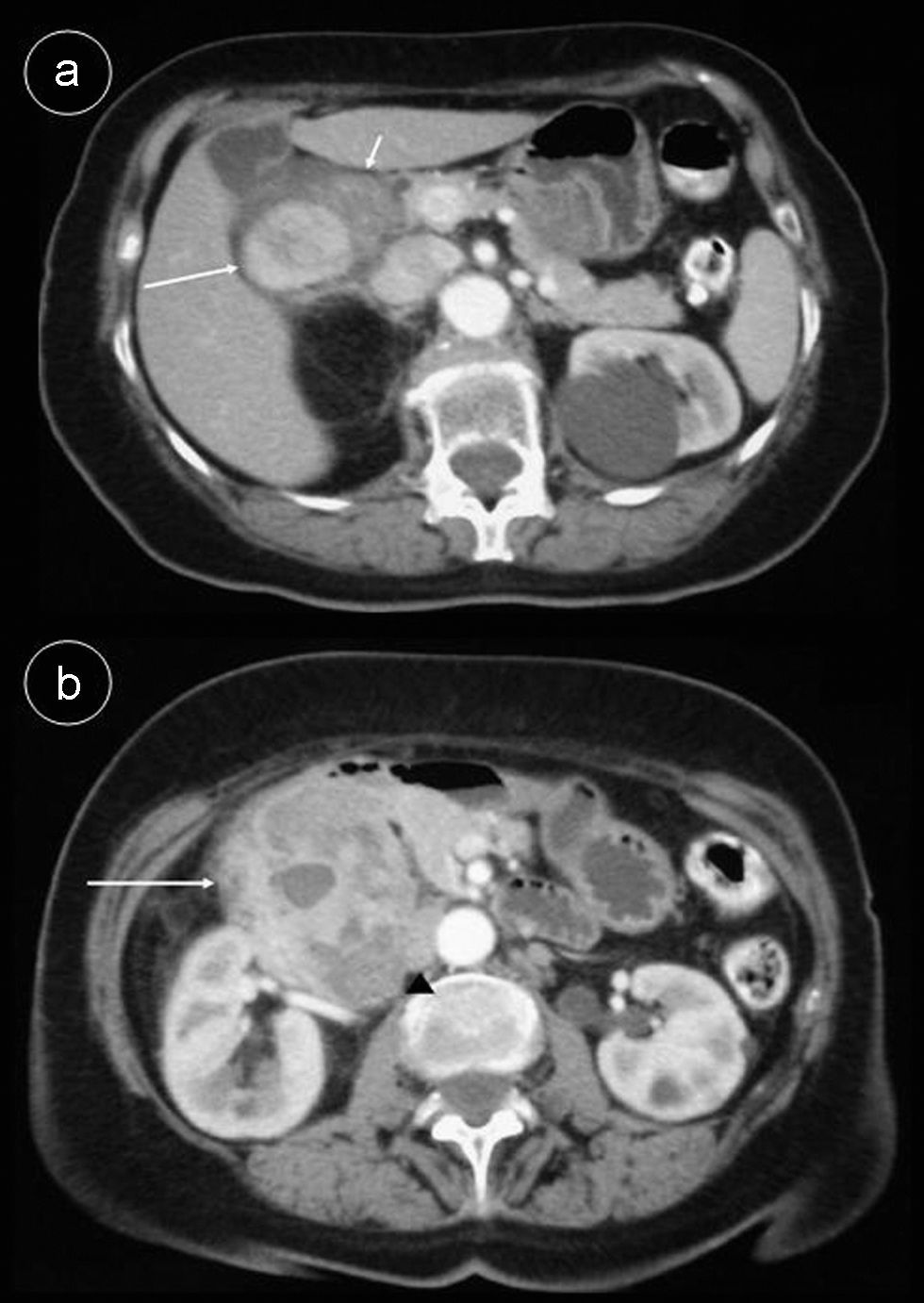
 Se visualiza la dilatación de la segunda porción duodenal (flecha corta) y el inicio de la neoplasia (flecha larga) con realce casi homogéneo. b) nivel inferior a a). La tumoración presenta un realce heterogéneo (flecha) desplazando sin invadirlos al proceso uncinado y la vena cava en sentido medial (punta de flecha).")
TC abdominal c/c en fase arterial. a) Se visualiza la dilatación de la segunda porción duodenal (flecha corta) y el inicio de la neoplasia (flecha larga) con realce casi homogéneo. b) nivel inferior a a). La tumoración presenta un realce heterogéneo (flecha) desplazando sin invadirlos al proceso uncinado y la vena cava en sentido medial (punta de flecha).
En esta paciente se realizaron controles mediante TC de tórax, abdomen y pelvis, detectándose a los 3 años recidiva local del tumor con una posible metástasis pulmonar, el estado general de la paciente empeoró drásticamente y muriendo antes de ser reintervenida. No se solicitó necropsia.
Caso 2Hombre de 55 años sin antecedentes de interés que consulta por dolor lumbar sin otra sintomatología; en el estudio de resonancia magnética (RM) de columna se apreció masa retroperitoneal hipointensa en secuencias potenciadas en T1 e hiperintensa en secuencias potenciadas en T2 (fig. 2), por lo que se indicó TC abdominal con contraste intravenoso (fig. 3), apreciándose en región pélvica una masa sólida, con bordes lobulados, bien definidos de 9,7×8×11cm, en cuyo interior se objetivaban áreas con intenso realce, alternadas con otras zonas hipodensas. La tumoración desplazaba sigma, vejiga y vasos iliacos hacia la derecha, sin infiltrarlos. Se realizó tumorectomía de forma programada. El estudio anatomo-patológico fue informado como tumor fibroso solitario tipo hemangiopericitoma. La tumoración estaba bien delimitada con aspecto de pseudocápsula, sin sobrepasar los límites de resección; presentaba abundante colágeno, aunque también se visualizaron células fusiformes con distribución asimétrica, objetivándose zonas hipercelulares con pleomorfismo celular y escasas mitosis. No se observaron áreas de necrosis. La inmunohistoquímica fue positiva para CD34, Bcl-2 y vimentina. Se demostró positividad nuclear para ki-67 en menos del 10% de las células neoplásicas, indicando una baja tasa de proliferación. Fueron negativos CD68, C-Kit, S-100, desmina y actina de músculo liso.
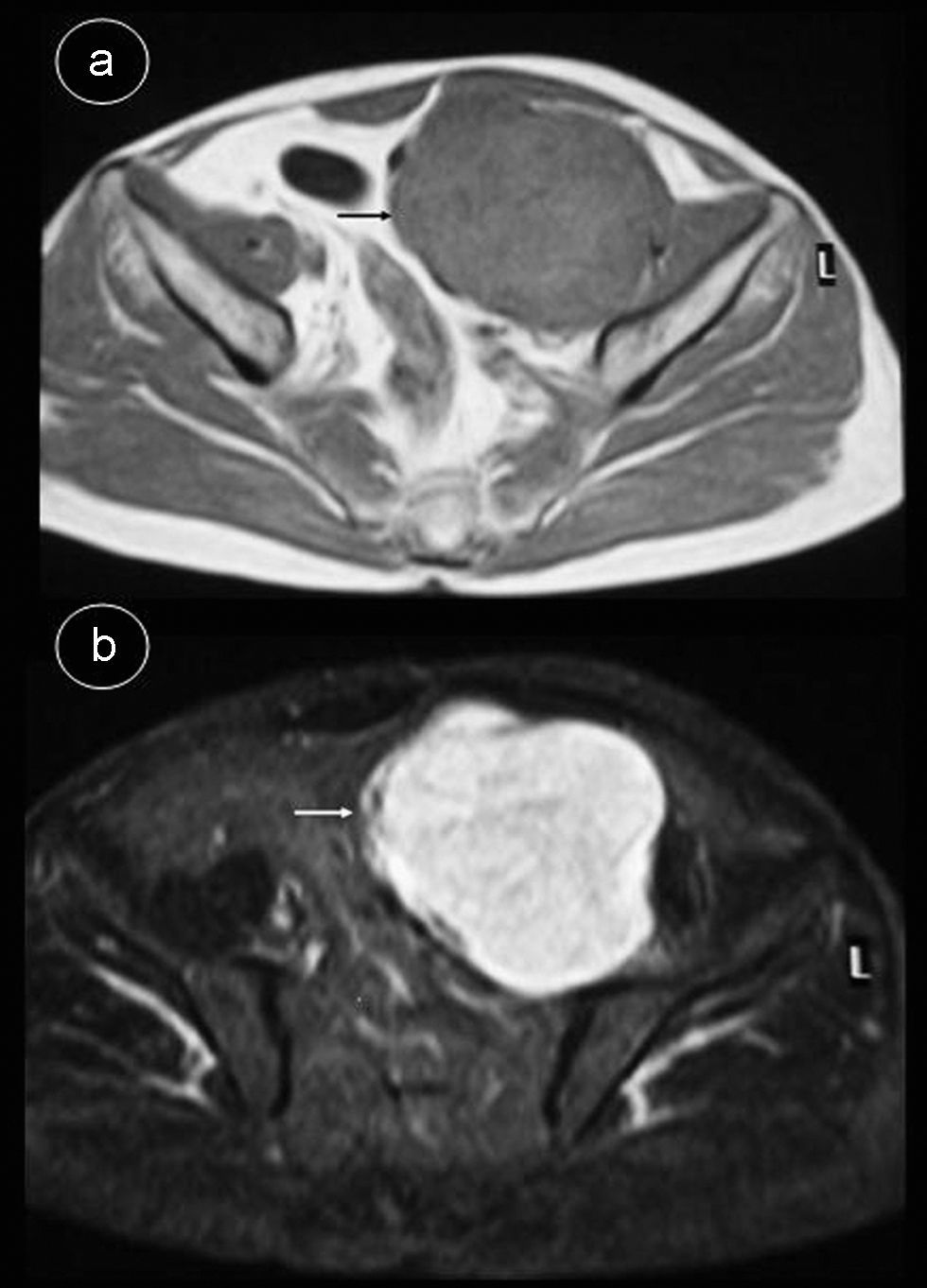
 Secuencia T1. Masa retroperitoneal isointensa con respecto al músculo (flecha). b) Secuencia T2. La tumoración es hiperintensa (flecha).")
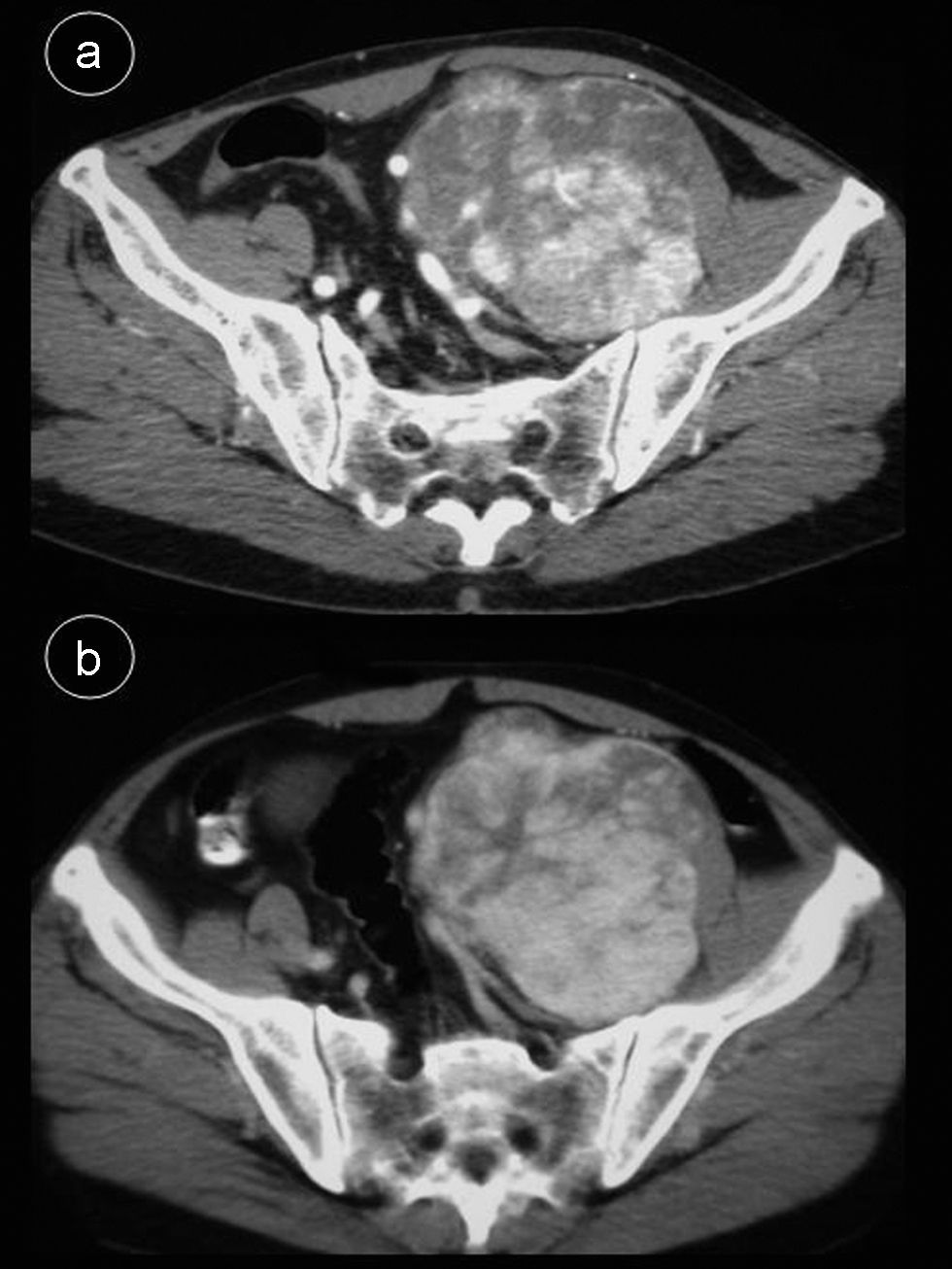
 Estudio en fase arterial. La tumoración presenta zonas con marcado realce precoz, que comienzan en la periferia. b) Mismo nivel de a). Fase portal. Se aprecia un aumento de las áreas realzadas hacia el centro del tumor alternándose con otras sin captación del contraste sugestivas de focos de colagenización.")
TC abdominal c/c. a) Estudio en fase arterial. La tumoración presenta zonas con marcado realce precoz, que comienzan en la periferia. b) Mismo nivel de a). Fase portal. Se aprecia un aumento de las áreas realzadas hacia el centro del tumor alternándose con otras sin captación del contraste sugestivas de focos de colagenización.
Al igual que el primer caso que presentamos el seguimiento oncológico de este paciente ha sido realizado mediante TC de tórax, abdomen y pelvis, sin que se hayan demostrado recidivas ni metástasis hasta la fecha, considerándose actualmente al paciente en remisión completa.
DiscusiónEn un inicio se describió el hemangiopericitoma como un tumor proveniente de los pericitos de Zimmerman1. No obstante, en la actualidad se acepta que el hemangiopericitoma tiene su origen en los fibroblastos; esto junto al hecho de que hasta un 15% de las neoplasias de partes blandas muestren un patrón de crecimiento de tipo hemangiopericitoide provocó su reclasificación; así, en la más reciente clasificación World Health Organization (WHO) de tumores de partes blandas,2 se propone el nombre de tumor fibroso solitario (TFS) con dos variantes: la celular, que resulta anatomopatológicamente indistinguible del hemangiopericitoma descrito por Stout y Murray en 1942 y la fibrosa, que se correspondería con el tumor fibroso solitario convencional,3 descrito por primera vez en la pleura por Wagner en 1870, que puede crecer además en cavidad abdominal y retroperitoneo, extremidades, tronco, cabeza y cuello, al igual que la variante celular puede alcanzar un gran tamaño y la sintomatología se produce principalmente por compresión de estructuras vecinas, no obstante a diferencia de ésta en el estudio anatomopatológico presenta una mayor abundancia de áreas de fibrosis.
El TFS variante celular afecta predominantemente a adultos sin predilección de sexo, con edades al diagnóstico que oscilan entre los 20 y 70 años; se localiza predominantemente en miembros inferiores y retroperitoneo, con menor afectación de cabeza y cuello, meninges, extremidades superiores o abdomen1,4; en nuestros casos las edades fueron superiores a 55 años y ambos se localizaban en retroperitoneo.
La mayor parte de los pacientes presentan síntomas derivados del crecimiento del tumor y la compresión que provoca sobre las estructuras vecinas, siendo en general una masa poco dolorosa; estas manifestaciones clínicas suelen ser insidiosas y preceder al descubrimiento del tumor que tiene un lento crecimiento1,4. En nuestros casos, el síntoma predominante fue dolor localizado, asociado a síndrome constitucional en el caso 1.
El diagnóstico se basa en el estudio anatomopatológico, sin embargo las técnicas de imagen pueden ayudar a sospechar la naturaleza del tumor debido a su gran vascularización además de estudiar sus relaciones con las estructuras vecinas permitiendo una mejor planificación del acto quirúrgico. En ecografía se visualiza como una masa bien definida, de ecogenicidad heterogénea e hipervascularizada en modo Doppler5,6. En la TC con contraste intravenoso, se aprecia una masa de partes blandas con márgenes definidos e hipervascularizada; la presencia de áreas de baja atenuación sugestivas de necrosis, calcificaciones distróficas e invasión de las estructuras vecinas pueden indicar un comportamiento maligno de la neoplasia5. En nuestros casos, las neoplasias presentaron un realce heterogéneo, correspondiendo las zonas de intenso realce a hipervascularización y las zonas hipodensas a colágeno solo (caso 2) o asociado a necrosis (caso 1). La RM suele comportarse isointenso al músculo en imágenes potenciadas en T1 con intensidad de señal variable en imágenes potenciadas en T2 (al igual que el caso 2)6,7. La captación de contraste es heterogénea en función del predominio de las áreas hipercelulares, los espacios vasculares, la matriz colágena y las posibles zonas quísticas7. En la angiografía suele revelarse como una masa muy hipervascularizada con opacificación centrípeta desde la fase arterial precoz, visualizándose un marcado aumento en el número y calibre de los vasos tumorales5,8.
El estudio anatomopatológico muestra una profusión de elementos vasculares, rodeados de células fusiformes de distribución asimétrica, con áreas hipercelulares y otras zonas hipocelulares con predominio de las fibras de colágeno y reticulina con dominancia de unas u otras según la variante celular o fibrosa de la neoplasia. Inmunohistoquímicamente suelen presentar positividad para CD34 (80–90%), CD99 (70%), Bcl-2 (30%), el antígeno epitelial de membrana (30%) y actina de músculo liso (20%). Suelen ser negativos para desmina, S-100 y las citoqueratinas. La positividad para CD34 es menos frecuente y fuerte en la variedad celular. Nuestros casos se comportaron de manera semejante.
El tratamiento de elección es la extirpación quirúrgica amplia. Puede realizarse una embolización selectiva previa a la cirugía para reducir el riesgo de sangrado masivo. La radioterapia y la quimioterapia no han demostrado su efectividad como tratamiento primario, aunque se han empleado como adyuvantes a la cirugía, en tumores irresecables y en recurrencias, con resultados desiguales. Actualmente se investiga el posible papel de la terapia antiangiogénica con temozolomide y bevacizumad dada la profusa vascularización del tumor7.
El pronóstico de estos tumores es bueno cuando el tratamiento ha sido potencialmente curativo, consiguiendo porcentajes de supervivencia a los 5 y 10 años del 80% y 70% respectivamente4. Sin embargo, aproximadamente un 15–20% de los pacientes con TFS muestran un comportamiento maligno consistente en recurrencias locales y metástasis a distancia a pesar del tratamiento quirúrgico. No existe certeza a la hora de predecir que casos tendrán un comportamiento más agresivo que otros, no obstante se han propuesto algunos criterios como: un gran tamaño (mayor de 6,5cm), invasión de estructuras vecinas, elevada actividad mitótica (más de 4 mitosis por 10 campos de gran aumento), presencia de hemorragia o necrosis y atipia celular9. A pesar de dichos criterios no existe ningún dato fiable para distinguir los TFS malignos, por ello se recomienda el seguimiento a largo plazo de estos pacientes ya que algunas recurrencias ocurren de manera tardía, incluso 10 años después de la neoplasia primaria. Las metástasis suelen asentar en pulmón seguidas de hueso e hígado4,8. El tratamiento de elección de las recidivas locales y metástasis, siempre que sea posible, es la extirpación quirúrgica1.
En conclusión, el TFS debe ser considerado en el diagnóstico diferencial de las neoplasias de partes blandas, hipervascularizadas que se originen principalmente en pelvis, retroperitoneo y miembros inferiores.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

