




El tumor fibroso solitario (TFS) es una rara neoplasia mesenquimal, de estirpe fibroblástica, que representa menos del 2% de todos los tumores de tejidos blandos1,2. Originalmente se describió en la pleura, e inicialmente se consideraba un tumor asociado a serosas1; sin embargo en los últimos años se han documentado casos procedentes de virtualmente cualquier localización anatómica y órgano3; en la actualidad se estima que el origen extrapleural es más frecuente1,4. Presentamos un caso de TFS abdominal, dependiente de la curvatura menor gástrica, sin ninguna sintomatología asociada y que fue hallado de forma casual al realizar una tomografía computarizada (TC) abdominal por otro motivo.
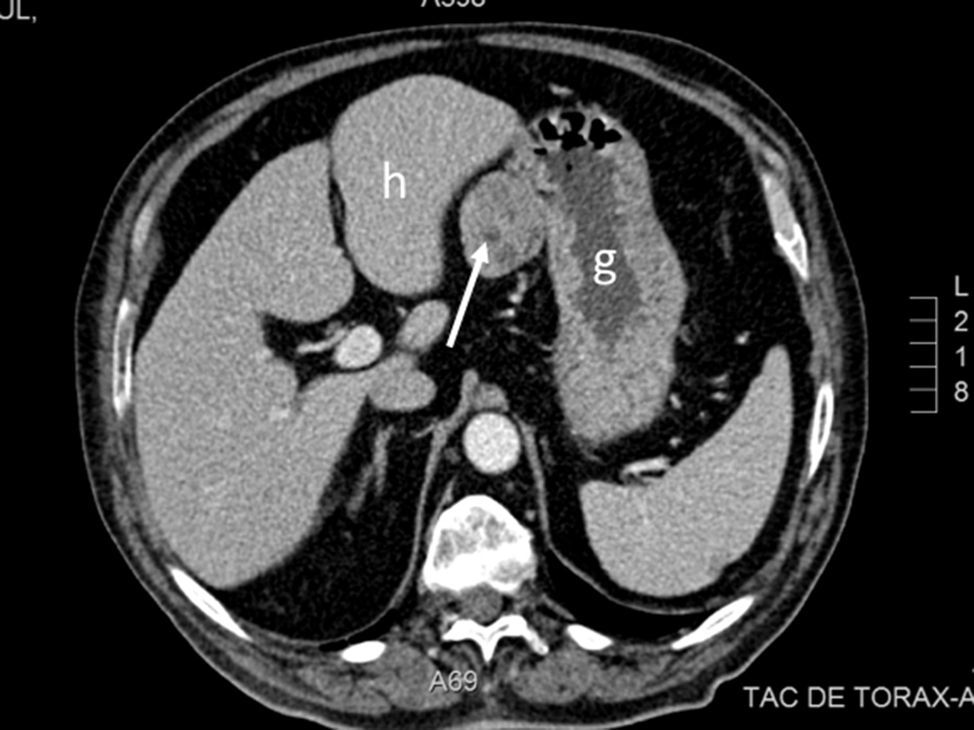
Se trata de un varón de 69 años con antecedentes de diabetes mellitus tipo 2, hipertensión arterial, cardiopatía isquémica, enfermedad pulmonar obstructiva crónica y síndrome de apnea del sueño, en tratamiento con metformina, glimepirida, sitagliptina, ácido acetilsalicílico, torasemida, eplerenona, bisoprolol, enalapril y valsartán. Refería un cuadro de varios meses de evolución de diarrea, astenia y pérdida ponderal. La exploración física no mostró alteraciones significativas. Se realizaron gastroscopia y colonoscopia, que no pusieron de manifiesto hallazgos relevantes. Se realizó, además, una TC abdominal en la que se observaba una masa sólida, homogénea, de 4,3cm de diámetro mayor, situada en el ligamento gastrohepático y en relación con la curvatura menor gástrica (fig. 1). Con la sospecha de tumor del estroma gastrointestinal (GIST) resecable se programó cirugía del mismo. En la intervención se observó una tumoración en la curvatura menor gástrica, con protrusión hacia la hoja anterior del epiplón menor. Se realizó resección laparoscópica, incluyendo escisión en huso del espesor completo de la pared gástrica sin incidencias reseñables. El estudio histológico mostró una tumoración de 6×5×4,5cm, bien delimitada, constituida por una proliferación homogénea de células fusiformes con patrón arremolinado, discreta atipia celular, ocasionales figuras de mitosis, así como estroma fibroso y abundantes estructuras vasculares con hialinización perivascular, compatible con TFS. Los márgenes de resección estaban libres de infiltración tumoral. El índice mitótico fue inferior a 2 mitosis/10 campos de gran aumento y el índice proliferativo bajo (Ki-67 del 1%). El estudio inmunohistoquímico mostró positividad para CD34, vimentina y Bcl-2, y negatividad para CD99, calretinina, c-Kit, DOG-1, D2-40, S-100, desmina y actina. El postoperatorio transcurrió sin complicaciones.
 situada entre el hígado (h) y el estómago (g).")
En la clasificación actual de los tumores de partes blandas el término TFS engloba a los hemangiopericitomas, así como al TFS propiamente dicho, considerados antes como entidades independientes5,6. Es una neoplasia poco común, de estirpe fibroblástica y que afecta a adultos entre 20-70 años. Histológicamente se caracteriza por zonas hipo o hipercelulares, con células fusiformes rodeadas de zonas de fibrosis y estroma colágeno, y un patrón vascular con vasos ramificados. En la inmunohistoquímica suele observarse positividad para CD34 y, con menor frecuencia, para CD99 y Bcl-23,5. Recientemente se ha establecido que el TFS se caracteriza por la fusión de los genes NAB2 y STAT6 del cromosoma 12, que conduce a la sobreexpresión de STAT6, a su activación y migración al núcleo celular. Esta alteración probablemente constituye el mecanismo patogénico inicial que explica el desarrollo tumoral. Por otra parte, se ha observado una alta sensibilidad y especificidad diagnósticas de la positividad nuclear para STAT6 en la inmunohistoquímica3,6.
Aunque inicialmente el TFS se consideró una tumoración de origen pleural, comunicaciones posteriores establecieron que podía asentar en cualquier órgano, incluyendo peritoneo, retroperitoneo, hígado, pulmón, estómago, vejiga y próstata; así mismo puede afectar a estructuras de cabeza y cuello, como parótida, tiroides, órbita y cavidad oral, tejido celular subcutáneo y, más raramente, piel3. Actualmente, se considera que el 60-70% de los casos tienen un origen extrapleural4,7. En las series más amplias destaca por su frecuencia la localización abdominal en un 30% de los casos4,7; sin embargo, el origen gástrico o en ligamento gastrohepático es excepcional8,9.
El TFS generalmente es un tumor localizado y benigno, aunque en un 15-20% de los casos exhibe un comportamiento agresivo que conduce a invasión local o enfermedad metastásica1; los factores que se han asociado a esta evolución son la edad, el tamaño tumoral y la presencia de un índice mitótico alto, atipia celular, necrosis, hemorragias o un borde tumoral infiltrativo1,4,5. Recientemente se ha elaborado un modelo de estratificación del riesgo basado en la edad, tamaño tumoral e índice mitótico4.
El tratamiento de elección del TFS es la extirpación quirúrgica. La eficacia de radioterapia y de la quimioterapia convencional es muy limitada. El abundante componente vascular de estos tumores sugiere que los agentes anti-angiogénicos pueden ser de utilidad1; por otra parte; la comprensión de las bases de la génesis tumoral (fusión NAB2-STAT6) abre las puertas a la posibilidad de nuevos tratamientos dirigidos frente a estas dianas moleculares en los casos de enfermedad no resecable o avanzada3.

