




Los tumores carcinoides gastrointestinales (TCa-GI) surgen desde células del sistema neuroendocrino difuso localizadas en el tracto digestivo y representan más del 70% de todos los TCa de los seres humanos. En este trabajo se revisan los siguientes argumentos: 1) El perfil biológico de los TCa-GI (dibujo histopatológico, marcadores citoquímicos, alteraciones metabólicas, almacenamiento de neuroaminas y proteínas hormonales, comportamiento citodinámico y características biológicas en función del origen embriológico). 2) Las circunstancias etiológicas (factores hereditarios excepcionales, asociación de TCa gástricos con gastritis autoinmune, factores exógenos poco conocidos). 3) Aspectos patogénicos (mitogénesis persistente de células endocrinas asociada a hipergastrinemia, inactivación de algunos presuntos genes supresores de tumor, dudosa participación de oncogenes, acción autocrina de algunas proteínas estimuladoras de crecimiento celular).4) Las repercusiones de ciertos episodios fisiopatológicos (reacción desmoplástica peritumoral responsable del «efecto masa» sobre el tubo digestivo, el «rapto» del triptófano alimentario por parte de las células tumorales hacia una vía metabólica anormal, la fácil diseminación metastásica coexistente con una escasa agresividad tumoral, la liberación al torrente sanguíneo de productos secretores almacenados responsables del «síndrome carcinoide» y de algunos cuadros de hiperfunción endocrina). Conviene recordar que los TCa-GI representan sólo un segmento de los llamados tumores neuroendocrinos y, como tales, deben considerarse.
Gastrointestinal carcinoid tumors arise from cells of the diffuse neuroendocrine system localized in the digestive trace and represent more than 70% of all carcinoid tumors in humans.The present article reviews the following topics:1) The biological profile of these tumors (histopathology, cytokine markers, metabolic alterations, storage of neuroamines and hormonal proteins, cytodynamic behavior, and biological behavior according to embryological origin). 2) The etiological circumstances (exceptional hereditary factors, association of gastric carcinoid tumors with autoimmune gastritis,little-known exogenous factors). 3) Pathogenic aspects (persistent mitogenesis of endocrine cells associated with hypergastrinemia,inactivation of some putative tumor suppressor genes, the doubtful participation of oncogenes, autocrine action of some cellular growth-stimulating proteins).4) The repercussions of certain physiopathological events (peritumoral desmoplastic reaction causing the «mass effect » on the digestive tube, the «kidnapping» of dietary tryptophan by tumoral cells toward an abnormal metabolic pathway; the easy metastatic dissemination coexisting with low tumoral aggressivity, and the release into the bloodstream of stored secretory products leading to «carcinoid syndrome » and some endocrine hyperfunction syndromes. Finally,it should be remembered that gastrointestinal carcinoid tumors represent only a proportion of the neoplasms classified as neuroendocrine tumors.
Conocemos con el término un tanto ambiguo de «tumores carcinoides» (TCa)1–5 a un conjunto de neoplasias epiteliales de crecimiento lento y prolongadamente asintomáticas, a pesar de su fácil diseminación metastásica, que pertenecen a la abigarrada familia de los tumores neuro- endocrinos (TNE)6,7. Esta familia de tumores incluye procesos tan variados como los neuroblastomas originados en la cresta neural, los feocromocitomas y adenomas hipofisarios surgidos en glándulas endocrinas convencionales, los carcinomas del páncreas endocrino (insulinomas, glucagonomas, etc.) procedentes de los islotes de Langerhans de aquel órgano y, por último, los TCa nacidos a partir de las células del llamado «sistema neuroendocrino difuso»7–10. Estas células se encuentran dispersas por toda nuestra economía, aunque se ubican muy preferentemente en los espacios interepiteliales y subepiteliales del tracto gastrointestinal (GI) y del árbol bronquial10.
Los TCa fueron descubiertos por Lubarch, en 188811, calificados como «Karzinoides», es decir, «parecidos a carcinomas », por Oberndorfer, en 190712, y considerados como neoplasias de naturaleza endocrina por Gosset y Masson, en 191413. Bien entrado el siglo pasado, en 1954, Thorson et al14 describieron el llamado «síndrome carcinoide» (diarrea, rubefacción facial, respiración sibilante, insuficiencia cardíaca derecha y manifestaciones pelagroides) y dejaron claro que este cuadro clínico era la consecuencia de la liberación al torrente sanguíneo de algunos de los productos secretores que almacenaban los TCa.
Se trata de neoplasias raras, ya que sólo representan el 1% de todos los tumores malignos de los seres humanos. En torno al 70% asienta en el tubo digestivo, algo más del 25% en el árbol bronquial y menos del 5% en otros órganos o sistemas de nuestra economía. Muestran una discreta preferencia por el sexo femenino (55%) y un pico de máxima incidencia diagnóstica entre la quinta y la séptima décadas de la vida2,4,5,15.
A pesar de su rareza, los TCa tienen especial importancia en gastroenterología por varias razones: en primer lugar, porque más de dos tercios se localizan en este territorio; en segundo lugar, porque estos tumores son, junto con el síndrome de Zolliger-Ellison, los grandes protagonistas de la «patología endocrinológica del tubo digestivo» y, por último, porque representan un tercio de los tumores del intestino delgado y la mitad de las neoplasias apendiculares. Intentaremos resumir en este trabajo la biología celular, la expresión molecular y las consecuencias fisiopatológicas de una neoplasia enigmática, como son los TCa-GI.
PERFIL BIOLÓGICO DE UNA NEOPLASIA MALIGNA «A CÁMARA LENTA»A lo largo y ancho de varios apartados trataremos de recoger los datos biológicos más característicos de los TCa, en general, y de sus formas GI, en particular1,3–5,16,17.
Descripción histopatológicaLos TCa-GI suelen aparecer como lesiones nodulares sólidas, únicas o múltiples, raras veces ulceradas. Estos tumores están habitualmente formados por capas monótonas de células pequeñas, uniformemente redondas, sin apenas atipias nucleares y escasas mitosis, que adoptan diversos patrones de crecimiento: insular (en nódulos sólidos), trabecular (en cintas entrelazadas), raras veces con disposición glandular o con aspecto indiferenciado y frecuentemente con un patrón mixto1,3,5,18.
Dos hechos histológicos, además de los descritos, destacan en estos tumores: por una parte, su acusada vascularización y, por otra, la intensa reacción desmoplástica que originan, en forma de fibrosis peritumoral. A pesar del patrón histológico de neoplasia indolente que coincide con su comportamiento clínico, y que mereció hace un siglo el calificativo de «carcinoide», hoy sabemos que en una pequeña fracción de los TNE-GI (menos del 20%) se aprecian signos histológicos de agresividad (displasia celular, índices proliferativos elevados, imágenes de angioinvasión y necrosis tumoral), que no siempre casan bien con su evolución patocrónica5,19.
El hallazgo ultraestructural en el citoplasma de las células carcinoides, de vesículas grandes (de más de 80 nm de diámetro), con un grueso gránulo electrodenso, donde se albergan los productos de secreción de aquéllas (aminas bioactivas y péptidos hormonales), junto con otras vesículas pequeñas (de 40-80 nm de diámetro), que corresponden a estructuras sinápticas neuronales, son 2 imágenes que avalan morfológicamente la naturaleza neuroendocrina de dichas células3.
Marcadores histoquímicosLa tinción de una parte de las células de los TCa-GI con cromato potásico justifica su definición como células «enterocromafin-símiles» (EC-s). Por otra parte, el contenido granular de las células carcinoides reacciona de manera diferente frente a las sales de plata. En una parte de dichas células este contenido reduce directamente las sales de plata (reacción argentafin), mientras que en otras, para que se produzca esta reducción hasta plata metálica es necesaria la intervención de un agente reductor exógeno (reacción argirófila)1,3,5.
Finalmente, con el tiempo se ha ido identificando una serie de proteínas ligadas a estructuras citoplasmáticas carcinoides que parecen comportarse como «marcadores paneuroendocrinos », más o menos específicos, entre los que se encuentran la enolasa específica de neuronas, la sinaptofisina y, sobre todo, las cromograninas A, B y C3–5,20.
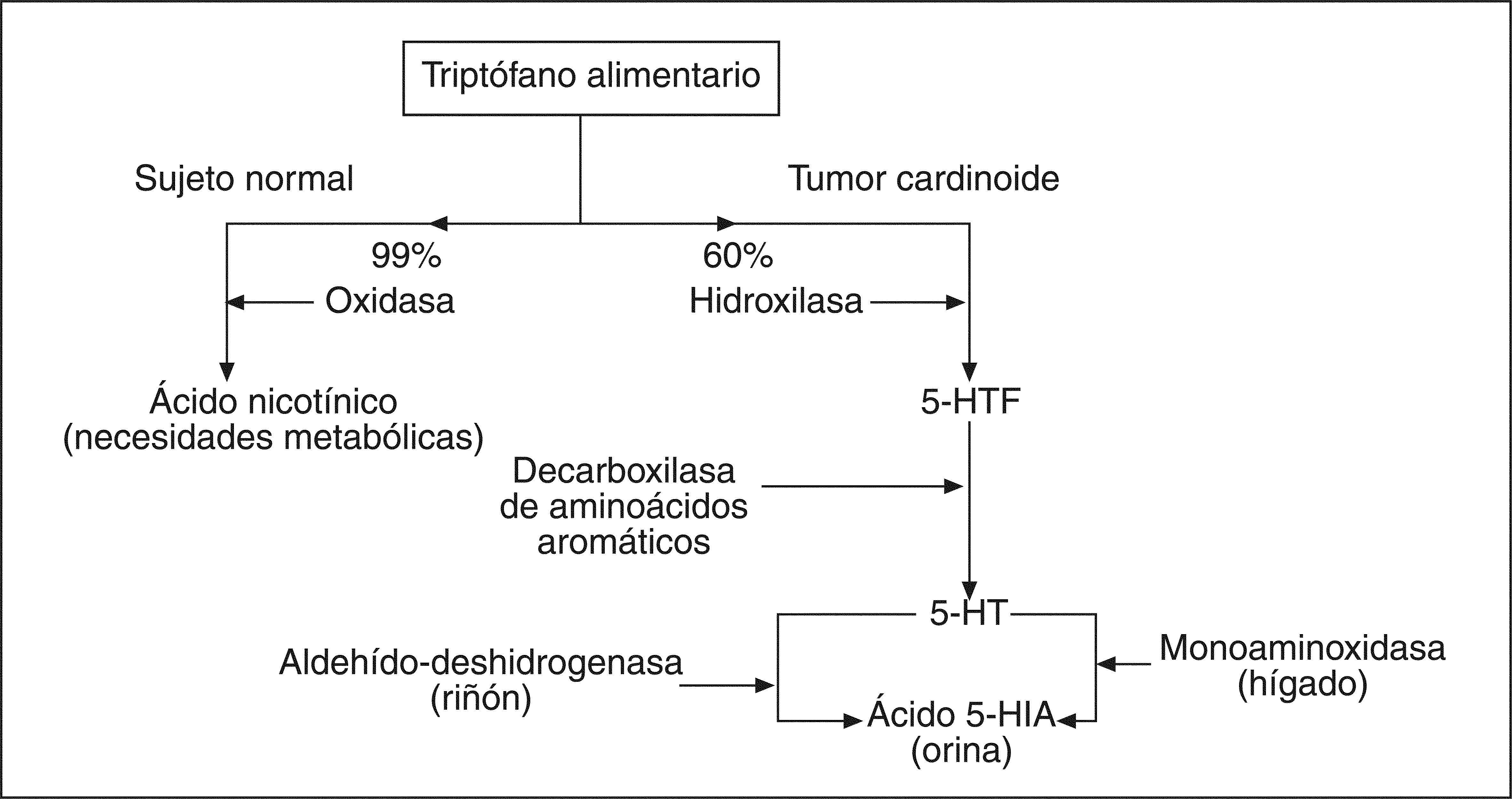
Alteraciones metabólicasPoco sabemos sobre las alteraciones metabólicas que presentan las células de los TCa-GI, con la excepción de la distorsión del metabolismo del triptófano, posiblemente originada por la presencia de muchos de estos tumores. Las cosas ocurren como a continuación comentaremos y resumimos en la figura 14,5.

Así, el triptófano alimentario, en condiciones fisiológicas, se metaboliza en el 99% de su aporte gracias a la acción de una oxidasa que lo transforma en ácido nicotínico, necesario para el metabolismo cerebral y de otros tejidos. Sólo un 1% de aquél sufre su transformación a 5-hidroxitriptófano (5-HTF) gracias a la acción de una hidroxilasa. Este modelo metabólico se rompe por completo, en una notable proporción de sujetos afectados de un TCa-GI, ya que las células de estos tumores «reclaman» para sí hasta el 60% del triptófano dietético, que es desviado hacia una hidroxilación de éste que lo transforma masivamente en 5-HTF. Este último metabolito, bajo la acción de una decarboxilasa de aminoácidos aromáticos, se transforma en 5-hidroxi-triptamina (5-HT) o serotonina que, a su vez, se degrada a ácido 5-hidroxi-indol-acético (Ac, 5-HIA). Este último paso metabólico se realiza fundamentalmente en el hígado gracias a la acción de la monoaminoxidasa y, en menor cuantía, en el riñón, gracias a la aldehído-deshidrogenasa. Por último, el ácido 5-HIA se elimina en la orina como producto metabólico terminal.
Esta distorsión del metabolismo del triptófano explica el anormal almacenamiento, en los gránulos electrodensos, de las células de muchos de estos tumores de 5-HT y 5-HTF. A título de epílogo cabría comentar que quizá algún TCa podría ser deficiente en la enzima decarboxilasa de ácidos aromáticos, por lo que quedaría bloqueado el anormal metabolismo del triptófano, eliminándose por el riñón tasas elevadas de 5-HTF en lugar de ácido 5-HIA.
Almacenamiento de productos de secreciónCon el paso del tiempo se ha ido descubriendo que, además de la 5-HT y del 5-HTF, las células de los tumores carcinoides son capaces de sintetizar, almacenar y, a veces, segregar unos 40 productos pertenecientes a diversas familias moleculares6. Entre éstas destacan las siguientes: aminas bioactivas (5-HT, 5-HTF, histamina, etc.); péptidos hormonales (ACTH, hormona del crecimiento, gastrina, glucagón, somatostatina, gonadotropina coriónica, calcitonina, etc.); taquicininas (sustancia P, neuropéptido K, kalicreína, etc.) y algunas protaglandinas (PG-E). La acción combinada de varias de estas sustancias, cuando llegan a la circulación general, conduce al desarrollo del denominado «síndrome carcinoide»1,5.
Receptores de membranaOtra característica del perfil biológico de las células de los TCa es la de expresar, en sus membranas, receptores para ciertas hormonas, como la somatostatina (SST-r), y varios factores de crecimiento, como el fibroblástico (FGF-r) y el derivado de las plaquetas (PDGF-r), etc. Estos receptores parecen desempeñar algún papel en la patogenia de estos tumores y en sus posibilidades diagnósticas y terapéuticas1,4,21.
Comportamiento citodinámicoLas células de la gran mayoría de los TCa-GI muestran una actividad proliferativa escasa con un índice mitótico exiguo y una tasa muy baja de células ki-67 positivas o PCNA (proliferating cell nuclear antigen) positivas. Esta débil capacidad multiplicativa con escasa citoagresión local contrasta con una notable facilidad para «exportar» fuera del tumor inicial a sus anodinas células neoplásicas, en acontecimientos metastásicos ganglionares o a distancia (hígado, huesos, etc.), en cuanto el tumor original excede de 1-2 cm de diámetro. A pesar de esta notable capacidad de diseminación y la escasa respuesta al tratamiento radioterápico, quimioterápico o inmunoterápico, la supervivencia a largo plazo es sensiblemente mejor que la de los adenocarcinoides gastrointestinales convencionales1,4. El comportamiento citodinámico de la mayoría de los TCa recuerda mucho al de los linfomas no hodgkinianos de células pequeñas, que suelen tener un «bajo grado» de agresividad, con una tendencia a la diseminación sistémica precoz, ganglionar y extraganglionar.
CARACTERÍSTICAS DE LOS TUMORES CARCINOIDES EN FUNCIÓN DE SU ORIGEN EMBRIONARIOSe han propuesto diferentes clasificaciones de los TCa basadas en su morfología18, su posible estratificación pro- nóstica19 o simplemente, en el caso de los TCa-GI, su distribución topográfica5. Sin embargo, la más conocida y quizá la más imaginativa fue la primera clasificación, propuesta por Williams y Sandler22 en 1963, basada en el origen embrionario de los espacios titulares en los que surgen. Y esto es así, en la medida en que, a pesar de sus defectos, intenta dibujar un perfil biológico diferenciado para las 3 áreas del intestino embrionario de las que procede la gran mayoría de territorios en los que surgen los TCa4,5,23.
Tumores carcinoides procedentes del intestino cefálicoDe esta región embrionaria procede el amplio grupo de los TCa bronquiales, que no es objeto de este trabajo, los TCa gástricos, duodenales y los raros casos de localización en el timo y las vías biliares. Todos juntos representan algo más del 30% de todos los TCa de nuestra economía. Considerados en conjunto, podemos decir que la gran mayoría de estas neoplasias son argirófilas, pero suelen comportarse como argentafin-negativas; acumulan, como productos secretores prominentes, el 5-HTF y la histamina, es escasa la capacidad de secreción de 5-HT, pero a veces liberan ACTH, hormona del crecimiento o gastrina.
Alrededor de un 5% de todos los TCa-GI surgen en el estómago a partir de las células EC-s situadas en su mucosa. Dentro de éstos cabe distinguir 3 tipos diferentes24–26. El tipo I, que representa el 75% de todos los TCa gástricos, se asocia etiológicamente a una gastritis crónica autoinmune (con o sin una anemia perniciosa desarrollada); en la mitad de los casos, este carcinoide da la cara con múltiples lesiones polipoides tumorales. El tipo II, que representa el 5-10% de todos los TCa gástricos, surge en sujetos afectados del síndrome multitumoral congénito, conocido como «neoplasias endocrinas múltiples tipo 1» (NEM-1) y se asocia frecuentemente a un síndrome de Zollinger-Ellison, por gastrinoma pancreático o extrapancreático. Por último, el tipo III es un TCa gástrico esporádico que representa el 20% de los carcinomas de esta localización. Sólo un 3% de los TCa-GI se localizan en el duodeno.
Tumores carcinoides procedentes del intestino medioAlgo más del 55% de todos los TCa de los seres humanos se desarrolla en territorios procedentes de este segmento embrionario del intestino. Entre ellos están los que se localizan en el yeyuno-íleon, el apéndice, el hemicolon derecho y algunos órganos extradigestivos, como los testículos y los ovarios.
El 40% de todos los TCa-GI surgen a partir de células endocrinas de situación intraepitelial del tracto yeyuno-ileal del intestino delgado; la inmensa mayoría de ellos se sitúa en el íleon y un tercio de estos últimos se manifiesta como lesiones carcinoides múltiples.
En torno al 23% de todos los TCa-GI surgen a partir de células endocrinas subepiteliales del apéndice, mientras que alrededor del 7% de los TCa-GI se localizan en el hemicolon derecho y surgen a partir de sus células endocrinas intraepiteliales.
La gran mayoría de los TCa procedentes de este segmento intermedio del intestino embrionario son argentafinpositivas y suelen segregar 5-HT y taquicininas (como la sustancia P), razón por la cual casi todos los TCa que se expresan clínicamente con el cuadro denominado «síndrome carcinoide» pertenece a este grupo.
Tumores carcinoides procedentes del intestino caudalEste grupo representa algo más del 15% de todos los TCa de los seres humanos, y se presenta en el hemicolon izquierdo, el recto y, raramente, el tracto genitourinario. En el grupo concreto de los TCa-GI, un 4% surge en el hemicolon izquierdo y un 18% en el recto, y lo hacen desde células endocrinas intraepiteliales. Se trata de tumores argentafin-negativos, en un 75% de los casos, y argirófilos, en un 55%, apenas muestran capacidad secretora y sólo en ocasiones liberan 5-HT y/o ACTH.
IMPACTO DE UNA ETIOLOGÍA OSCURAEn la gran mayoría de las neoplasias malignas gastrointestinales podemos diferenciar, fundamentalmente, 3 grupos de circunstancias etiológicas posibles, a saber: factores genéticos hereditarios, factores endógenos no genéticos y factores exógenos (alimentarios, tóxico-ambientales, etc.). Cabría especular con un cuarto factor al que podríamos denominar «error por azar», al que toda célula con capacidad multiplicativa se encuentra expuesta27–29.
Factores genéticos hereditariosUna historia familiar de los TCa se ha comunicado en menos del 1% de los casos, y se sabe que el riesgo de desarrollar un tumor de estas características por parte de familiares de primer grado de un sujeto con un diagnóstico en firme es 3 veces superior a lo normal3.
Sin embargo, las formas hereditarias de TCa prácticamente sólo se descubren en sujetos afectados del síndrome hereditario conocido como NEM-1. Este cuadro se caracteriza por una predisposición familiar a desarrollar adenomas paratiroideos, hipofisarios y de los islotes de Langerhans pancreáticos, entre otros1–5,30,31. Como es sabido, se debe a una inactivación por mutación/deleción de los alelos del gen NEM-1, situado en el cromosoma 11q1.3; algo más del 20% de los casos afectados de este síndrome multineoplásico presenta también un TCa fundamentalmente gástrico (tipo II), y la mitad de los pacientes con un síndrome NEM-1 presenta igualmente un síndrome de Zollinger-Ellison por gastrinomas múltiples (pancreáticos o extrapancreáticos) concomitantes.
Se ha comunicado, con carácter excepcional, la asociación de algún caso de TCa duodenal con la neurofibromatosis tipo 13, originado por una mutación del gen supresor de tumor NF-1 situado en el cromosoma 17q1.1.
Factores endógenos: gastritis autoinmuneUna circunstancia etiológica que afecta al 75% de los TCa-GI (los pertenecientes al tipo I) es el desarrollo de una gastritis autoinmune crónica (con o sin anemia perniciosa). Este acontecimiento autoagresivo es responsable de un 3-4% de todos los TCa-GI1,3,5,25,26,30.
Otros factores (¿exógenos?)En algo más del 10% de los TCa-GI coexisten, con carácter sincrónico o metacrónico, estructuras neoplásicas adenocarcinomatosas no endocrinas con otras claramente neuroendocrinas15,32,33. Esta «colisión tumoral» de 2 fenotipos celulares diferentes se ha encontrado con mayor frecuencia en los cánceres colorrectales.
La coexistencia de patrones neoplásicos neuroendocrinos «incrustados» en estructuras adenocarcinomotoras indica que probablemente las mismas circunstancias etiológicas (¿exógenas?) que ponen en marcha la tumorigénesis adenocarcinomatosa deben ser las que hagan otro tanto con el sustrato neuroendocrino. Aunque, a decir verdad, poco sabemos sobre la razón íntima de esta «bivalencia neoplásica ».
ASPECTOS RELEVANTES DE LA PATOGENIAComo acabamos de mencionar, las circunstancias etiológicas de los TCa-GI son prácticamente desconocidas en más del 95% de los casos. Nada tiene de particular que ocurra algo parecido en las vías patogénicas de los factores etiológicos, conocidos o desconocidos, para realizar estas neoplasias del «sistema neuroendocrino difuso» del tubo digestivo.
A continuación comentaremos, de manera un tanto desordenada, algunas parcelas patogénicas sobre las que tenemos cierta información10,34–36.
Mitogénesis persistente tras la gastritis autoinmuneEl mecanismo a través del cual una gastritis crónica autoinmune puede estimular las células EC-s del fundus y el cuerpo gástricos a proliferar en sentido oncogénico, en uno o varios puntos de su mucosa, podría guardar relación con la atrofia de ésta y la aclorhidria que causa dicho proceso y, tras ello, la reacción hipergastrinémica que tal situación conlleva2,10,25,36. Esta hipergastrinemia provocada por las células G del antro gástrico estimula la proliferación sostenida de las células EC-s gástricas, y con ello quizá se cumpla la ley no escrita de la tumorigénesis que dice que «todo lo que es persistentemente mitogénico termina siendo potencialmente mutagénico»37.
La importancia de esta vía patogénica se apoya en trabajos de patología experimental. Así, se ha comprobado que es posible desarrollar TCa-GI en ratas sometidas a tratamientos prolongados con inhibidores de la bomba de protones (IBP), en dosis elevadas38. Igualmente, se sabe que el roedor Mastomys desarrolla, en un 30-40% de los casos, un TCa-GI en los primeros 2 años de vida, acontecimiento que ocurre, en apenas 4 meses, cuando se le administra una terapéutica inhibidora de la secreción gástrica39. En ambos modelos experimentales se provoca una hipersecreción estimulante de gastrina, como probable mecanismo oncogénico.
Tanto en la tumorigénesis humana tras una gastritis autoinmune como en los modelos experimentales comentados, destaca la existencia de zonas con hiperplasia de células EC-s, en torno a las lesiones carcinoides. Se trata, probablemente, de estructuras precancerosas que, en cualquier momento, se pueden malignizar, con lo que podría explicarse el carácter frecuentemente multifocal de este tipo de TCa-GI40.
Sin embargo, a pesar de estos hechos, nunca se ha logrado demostrar con seguridad, en los seres humanos, la asociación de TCa-GI con la administración prolongada de IBP, como antiácidos, ni tampoco con la existencia de un síndrome de Zollinger-Ellison esporádico41. Estos últimos hechos sugieren que, en el hombre, es necesario «algo más» que una hipergastrinemia crónica para provocar una oncogénesis carcinoide gástrica; quizá algún factor de predisposición genética y/o el efecto concomitante de una agresión autoinmune o de otra naturaleza.
Cuando intentamos extrapolar este modelo patogénico de oncogénesis carcinoide a la región ileal del intestino delgado, donde también son relativamente frecuentes los TCa multifocales, tenemos que reconocer que desconocemos, hoy por hoy, el estímulo mitogénico que podría incitar a las células endocrinas interpiteliales a malignizarse en una o muchas lesiones carcinoides. Es, como mínimo, sorprendente, a este respecto, el hallazgo puntual de algún autor42 que, al estudiar la clonalidad de neoplasias carcinoides ileales múltiples, señala un patrón de inactivación del cromosoma X idéntico para todas las lesiones de un mismo paciente; este hallazgo sugiere que podríamos estar ante una diseminación metastásica intraepitelial.
Alteraciones genéticas en la tumorigénesis carcinoideComo se sabe, hay varias familias de genes, en la cadena de ADN nuclear de los seres humanos, que codifican, en condiciones normales, proteínas que modulan la proliferación, la diferenciación y la muerte celular programada (apoptosis), entre otros acontecimientos vitales. Estas es- tructuras nucleotídicas pueden sufrir, en las células germinales o somáticas, alteraciones genéticas (mutaciones, translocaciones recíprocas, deleciones o amplificaciones) o modificaciones epigenéticas funcionales (inactivación de un gen por una anormal mutilación de su segmento promotor) con claras implicaciones en la patogenia neoplásica. Las principales familias de genes, con posible participación en la tumorigénesis en general y en la de los TCa en particular, son: genes reparadores del ADN, genes supresores de tumor, protooncogenes y otros nucleótidos que codifican proteínas calificadas como «factores de crecimiento » y sus receptores43.
A continuación, intentaremos resumir lo que hoy conocemos sobre la participación de dichas familias en los TCa-GI.
Genes reparadores del ADNLa replicación del ADN es un acontecimiento posmitótico causado por la enzima ADN-polimerasa, que empareja adecuadamente las bases purínicas y pirimidínicas. Cuando esta función enzimática fracasa, se produce un peligroso emparejamiento inadecuado que inestabiliza el genoma con riesgo de acontecimientos mutacionales. Para evitar estos acontecimientos, hay toda una serie de genes (MLH1, MSH2, PMS1, PMS2, etc.) que codifican la síntesis de proteínas capaces de reconocer y corregir este «desparejamiento de bases»44.
Se supone que la inactivación de alguno de estos genes reparadores del ADN debe ser uno de los primeros acontecimientos de muchas secuencias oncogénicas, aunque por el momento no tenemos claras evidencias de que esto ocurra en los TCa-GI.
Genes supresores de tumorUna segunda familia de secuencias nucleotídicas del ADN cuya inactivación genética (por mutación/deleción) o epigenética (por supresión funcional tras la anormal metilación de los nucleótidos del fragmento promotor del gen) parecen claramente implicados en la carcinogénesis humana, la de los denominados «genes supresores de tumor». En condiciones fisiológicas, estos genes codifican la síntesis de proteínas cuya función, en términos generales, es frenar la entrada en ciclo nuclear de las células con algún tipo de lesión en su genoma y, si esto no es posible, propiciar su apoptosis. La mayoría de estos genes parecen comportarse como recesivos, por lo que la pérdida completa de su función protectora del genoma requiere su inactivación bialélica (genética o epigenética). Pertenecen a esta familia de genes los siguientes: p53, p16, p27, gen de la poliposis colónica familiar (APC), gen del retinoblastoma (Rb), etc.45.
Es bien conocida la participación de varios de estos genes en la patogenia de neoplasias del tracto digestivo, como el cáncer colorrectal, el cáncer gástrico y el cáncer del páncreas exocrino27–29. Entre ellos, merece especial mención el papel del gen p53, situado en el cromosoma 17q2.1, por su frecuente implicación en la oncogénesis de neoplasias digestivas. Sin embargo, la inactivación de este gen sólo se ha demostrado en aislados TNE pulmonares morfológicamente atípicos5,46,47 y no parece desempeñar ningún papel importante en los TCa-GI48. El presunto gen supresor de tumor Reg 1 alfa codifica la síntesis de una proteína segregada por las células EC-s y por las células principales gástricas que, con carácter autocrino o paracrino, inhibe la estimulación proliferativa de la gastrina sobre las células EC-s. La inactivación mutacional de este gen se ha implicado, por parte de algún autor, en la patogenia de la oncogénesis carcinoide posgastritis autoinmune49.
Mucha mayor importancia tiene la inactivación por mutación/ deleción de otro presunto gen supresor de tumor, como es el responsable del síndrome NEM-1, situado en el cromosoma 11q1.3. Como observamos en el apartado anterior, al menos un 20% de los pacientes con un síndrome NEM-1 presenta un TCa-GI tipo II, como parte del cuadro multineoplásico. La inestabilidad genética que provoca la inactivación de este gen y el efecto mitogénico en las células EC-s del fundus y el cuerpo gástricos que origina la hipergastrinemia del síndrome de Zollinger-Ellison, que suele acompañar a aquel acontecimiento genético, podría explicar la patogenia de esta subvariedad de TCa-GI tipo II50. La utilización en este contexto (síndromes NEM-1 y Zollinger-Ellison) de IBP, como los antiácidos, podría incrementar todavía más el riesgo de TCa-GI51,52.
Sin embargo, no parecen terminar aquí las cosas, ya que la posible inactivación del gen NEM-1 podría estar también implicada en la realización de TCa esporádicos, no sólo gástricos sino también pulmonares, es decir, de TCa originados en órganos derivados del intestino cefálico embrionario. Esta idea se ha reafirmado al estudiar, con diversas técnicas de genética molecular (ensayos para detectar pérdidas de heterocigosidad, técnicas de hibridación genómica comparativa, etc.), las muestras de material de estos tumores. Este tipo de estudios ha permitido poner de manifiesto las posibles deleciones en el cromosoma 11q (donde está situado el gen NEM-1) y, en ocasiones, mutaciones de éste, entre el 36 y el 70% de TCa broncopulmonares y gástricos esporádicos53–56. De todas maneras, conviene matizar que estas deleciones, en el brazo largo del cromosoma 11, podrían afectar no sólo a la región 11q1.3 del gen NEM-1, sino también a zonas vecinas (p. ej., 11q2.3)57. También se han encontrado deleciones en estos TCa, en casos aislados, en otros cromosomas, como 3p, 5q, 9p, 10q y 13q. Desconocemos si en éstos hay genes supresores de tumor implicados.
Aplicando la misma metodología genético-molecular al estudio de TCa surgidos en áreas derivadas del intestino medio embrionario (yeyuno-íleon, apéndice y hemicolon derecho), algunos grupos de investigadores58–61 han encontrado datos sugestivos de pérdidas de material genético en los cromosomas 18q en un 67-88% de los casos, 18p en el 43% de los tumores, 11q22 en el 33% de las muestras estudiadas, y 16q y 9p en el 22%3. En estas localizaciones podrían estar ubicados presuntos genes su- presores cuya deleción favorecería la tumorigénesis que nos ocupa.
Para terminar de complicar las cosas, estos estudios detectaron ganancias de material genético en los cromosomas 17q y 19p en un 57% de los tumores, 19q o 4q en el 50% de los casos, y 4p en un 22%. Desconocemos el significado íntimo de estas distorsiones genéticas «por exceso». Las diferencias en la localización principal de los presuntos genes supresores de tumor –11q en los TCa derivados del intestino cefálico y 18q en los derivados del intestino medio embrionarios–, en relación con otras alteraciones genéticas menos frecuentes, apuntan 2 hechos: por una parte, que las pérdidas genéticas en estas localizaciones son acontecimientos patogénicos precoces en la gestación de TCa originados en órganos derivados de ambas áreas embrionarias y, por otra parte, que ambas tumorigénesis, siguen vías patogénicas diferentes, aún no aclaradas.
Activación de protooncogenesUna tercera familia de estructuras nucleotídicas, que por su parecido con los oncogenes virales responsables de la capacidad tumorigénica de ciertos retrovirus recibieron la denominación de protooncogenes, puede estar implicada en la patogenia de numerosas neoplasias, después de sufrir determinados acontecimientos mutacionales. Se trata de genes que actúan con carácter dominante, de tal manera que el cambio estructural de una de sus copias (por mutación puntual, translocación recíproca, etc.) lo activa transformándolo en un encogén celular. El producto codificado por este oncogén es una proteína «con exceso de función» inductora de una proliferación celular anormal. A esta familia de oncogenes celulares pertenecen, entre otros muchos, los genes del grupo ras (K-ras, N-ras y Hras), myc (c-myc, n-myc), abl, fos, src, bcl-2, etc.62.
Así, se ha encontrado en TCa de ratones transgénicos, la activación de los oncogenes de acción nuclear c-myc, nmyc y c-jun63; también se ha detectado, en algunos TNE pulmonares humanos morfológicamente atípicos, una hiperexpresión de algunas oncoproteínas procedentes de oncogenes, como c-fos, c-jun, c-net y c-myc30,35,64. Sin embargo, ninguno de ellos parece claramente implicado en la realización de TCa-GI, salvo una amplificación del protooncogén HER-2/neu encontrado en algún TNE gastroenteropancreático65,66.
En el ámbito de la oncología molecular se sabe que los genes bax y bcl-2 codifican la síntesis de proteínas con una elevada analogía estructural, y modulan, en sentido opuesto, el fenómeno de la apoptosis que facilita el sano ejercicio de eliminar, por muerte programada, las células genéticamente anormales. La proteína bax facilita dicha apoptosis y, por tanto, se comporta como antitumoral; por el contrario, la proteína bcl-2 impide la apoptosis y se comporta como protumoral. Hay indicios de que en algunos TCa pulmonares predomina la expresión prooncogénica bcl-2 respecto a la antioncogénica bax67, y también se ha implicado a la oncoproteína bcl-2 en la fase inicial de algunas TCa-GI68.
Participación de factores de crecimientoDesconocemos cuál es la razón fundamental de la hiperexpresión, en ciertos TCa-GI, de algunas proteínas calificadas como «factores de crecimiento», cuya síntesis está codificada por genes que no se incluyen en la familia de los protooncogenes. Nos referimos al factor de crecimiento fibroblástico (FGF), el factor de crecimiento endoteliovascular (VEGF) y el factor de crecimiento epidérmico (EGF), así como los factores de crecimiento transformante alfa y beta (TGF-α y TGF-β). También se encuentran notablemente expresados algunos receptores de dichos factores, como el EGF-r, el FGF-r y el del factor de crecimiento derivado de las plaquetas (PDGF-r), entre otros21,69,70. Muchas de estas proteínas podrían colaborar, con carácter autocrino, a la realización tumoral carcinoide y quizás también, con carácter paracrino, en los acontecimientos fisiopatológicos que completan su realización.
REPERCUSIONES FISIOPATOLÓGICAS EN LA EXPRESIÓN CLINICOBIOLÓGICAA la transformación tumoral de las células del sistema neuroendocrino difuso, localizadas en la mucosa gastrointestinal, sigue una serie de acontecimientos fisiopatológicos, que terminan perfilando el cuadro clinicobiológico del proceso. A continuación comentaremos los acontecimientos mejor conocidos y sus consecuencias4,34,35,47.
Reacción desmoplásticaUn primer acontecimiento fisiopatológico es la intensa reacción desmoplástica que acompaña al lento crecimiento local de un TCa-GI, a expensas del tejido conjuntivo peritumoral. La acción paracrina de algunos factores de crecimiento (FGF, etc.) liberados por las células carcinoides parecen desempeñar un papel importante en este hecho, principal responsable del «efecto masa» en el tracto digestivo1,4,5,21. La expresión clínica de este «efecto masa» implica, según la localización y el tamaño del tumor, la aparición de disfagia, dolor abdominal, síntomas de obstrucción intestinal, hemorragias digestivas, cuadro apendicular, perforación, etc.
Diseminación de células poco agresivasEn segundo lugar, cabe destacar la gran facilidad con la que los TCa «exportan» a sus pequeñas y anodinas células fuera de la lesión original, en forma de metástasis a los ganglios locales y a distancia (hígado, huesos, etc.), en cuanto aquél sobrepasa el tamaño de 1-2 cm de diámetro. Es posible que la notable vascularización de esta neoplasia, realizada bajo el efecto autocrino de otro factor de crecimiento, como el VEGF, desempeñe algún papel en este acontecimiento que, como después veremos, propicia la aparición de síntomas secretores16,21,70–72.
Este tráfico celular que facilita el origen de las metástasis precoces contrasta con una evidente hipodinámica multiplicativa de la gran mayoría de los TCa-GI, que se comportan como neoplasias malignas «a cámara lenta». Así, la supervivencia global a los 5 años de los TCa del apéndice alcanza el 98% de los casos, los del recto el 87%, los gástricos tipos I/II el 81%, los del colon el 62%, los del intestino delgado el 60% y los TCa gástricos tipo III el 33%3.
Conviene recordar, a este respecto, que la coexistencia de un tumor de escasa agresividad celular y fácil diseminación sistémica se da también en otras neoplasias humanas, como los linfomas no hodgkinianos de bajo grado.
«Secuestro» del triptófano alimentarioUn tercer acontecimiento que desencadena consecuencias fisiopatológicas importantes es «el secuestro» que hacen las células carcinoides de una parte importante del triptófano alimentario, como observamos en un apartado anterior4,5. Este hecho conlleva dos consecuencias importantes (fig. 1). Por una parte, la acumulación intracelular de las aminas bioactivas más prominentes (como la serotonina o 5-HT y el 5-HTF) y la eliminación urinaria de cantidades elevadas del ácido 5-HIA, su metabolito terminal, sumamente útil para el diagnóstico de estos tumores. Por otra parte, esta desviación del triptófano dietético por la vía catabólica de la hidroxilasa puede provocar un déficit de ácido nicotínico necesario para algunas funciones metabólicas (cerebrales, cutáneas, etc.), responsable del cuadro pelagroide que presentan algunos pacientes16,47,72.
Liberación de productos secretores a la circulaciónUn acontecimiento trascendental que complica el desarrollo de bastantes TCa-GI es la entrada en la circulación sistémica de algunos de los productos que almacenan sus células, es decir, ciertas aminas bioactivas, algunos péptidos hormonales y determinadas taquicininas y prostaglandinas4,5,16,47.
Así, se sabe que la acción prolongada de algunas hormonas almacenadas en las células de algunos TCa y vertidas a la sangre (ACTH, hormona del crecimiento, gastrina, etc.) puede causar, en raras ocasiones, síndromes de hiperfunción hormonal (síndrome de Cushing, acromegalia, síndrome de Zollinger-Ellison, etc.).
Mucha mayor importancia tiene el efecto sinérgico que provoca la entrada en la circulación de ciertas aminas bioactivas (5-HT e histamina) y algunas taquicininas (sustancia P, etc.) y prostaglandinas (PG-E, etc.) en una parte significativa de pacientes, y que conocemos como «síndrome carcinoide». Este cuadro, en su forma completa, se expresa con diarrea, rubefacción cutánea, broncospasmo (sibilancias/asma), cardiopatía por lesión fibrótica de válvulas de cavidades derechas y manifestaciones cutáneas pelagroides. Este síndrome aparece generalmente en pacientes con TCa-GI surgidos en territorios derivados del intestino medio embrionario (yeyuno, íleon, apéndice y hemicolon derecho). En el 90% de los casos estos tumores han provocado metástasis hepáticas o, en su defecto, una amplia invasión retroperitoneal con acceso directo a la circulación sistémica. Sólo ocasionalmente, o de forma atípica, surge un síndrome carcinoide en casos situados en territorios procedentes de otros segmentos del intestino embrionario.
Expresión de un receptor de membrana de la somatostatinaLa somatostatina (SST) segregada por las células D del fundus gástrico, del duodeno y las células delta del páncreas endocrino, entre otras, actúa, en términos generales, como una hormona inhibidora de la secreción de las células endocrinas, efecto al que se suma una cierta actividad antiproliferativa1,4.
La riqueza de receptores de membrana para esta hormona (SST-r), en el 90% de los TCa, tiene algunas consecuencias fisiopatológicas. En primer lugar, quizá tenga algún papel en el perfil hipodinámico y relativamente quiescente de una neoplasia maligna, como es el TCa. Por otra parte, estos receptores permiten que un análogo de la SST, como la octreótida, marcado con un isótopo radiactivo (In111), ofrezca una imagen gammagráfica útil para el diagnóstico, y con un incremento de las dosis de octreótida- In111 es posible actuar terapéuticamente mejorando los síntomas secretores en un 60-80% de los pacientes, y obtener cierto grado de reducción tumoral en algunos casos (15-30%).
COMENTARIOS FINALESEl hecho que de que la actividad secretora endocrina no siempre vaya ligada a una disposición celular en amasijos glandulares se ha incorporado a las bases de la fisiología desde hace apenas un siglo.
Por otra parte, la existencia de un polifacético sistema neuroendocrino difuso de células dispersas por toda nuestra economía, aunque preferentemente ubicadas en la barrera epitelial o en los espacios subepiteliales del tracto gastrointestinal y del árbol bronquial, se contempla, aún hoy en día, como un acontecimiento enigmático. Este hecho convirtió al tubo digestivo en el principal órgano endocrino de los seres humanos y dio pie a la «era de la endocrinología gastrointestinal», sobre todo después de que en 1955 se describiese el síndrome de Zollinger-Ellison. La posibilidad que desde aquellos elementos neuroendocrinos dispersos puedan originarse neoplasias, solitarias o múltiples —que por su reducida agresividad recibieron el calificativo de «carcinoides» y que una parte de ellas se expresen con cuadros clínicos de hipersecreción de neuroaminas y péptidos hormonales— convierte a estas neoplasias en una fascinante realidad biológica.
No obstante, si consideramos los TCa desde una cierta altura conceptual, nos damos cuenta de que, en realidad, es- tas neoplasias sólo representan un «segmento» del amplio espectro biológico de los TNE, que no siempre se comportan como neoplasias malignas «a cámara lenta»19,47. Algunos autores han propuesto incluir a los TCa-GI dentro de un amplio paquete de neoplasias a las que denominan TNE gastroenteropancreáticos73. Aunque la tendencia semántica parece ir en esta dirección, quizá lo más razonable sería, de momento, llamarlos simplemente TNE seguidos de un «primer apellido» que haga referencia al segmento donde asientan y un «segundo apellido» que oriente sobre su presunta agresividad tumoral (bajo grado frente a alto grado).
De todas maneras, no será fácil borrar de la literatura médica un término como el de «tumores carcinoides», con el que se ha designado durante un siglo a un conjunto de neoplasias (únicas o múltiples) con una fuerte personalidad celular, molecular y clínica, como hemos intentado recordar en este trabajo.

