




Non-alcoholic fatty liver disease (NAFLD) is the most common liver disease in the world, with epidemiological studies indicating a 25% prevalence. NAFLD is considered to be a progressive disease that progresses from simple hepatic steatosis to non-alcoholic steatohepatitis (NASH), then to liver fibrosis, and finally to cirrhosis or hepatocellular carcinoma (HCC). Existing research has mostly elucidated the etiology of NAFLD, yet its particular molecular processes remain uncertain. Long non-coding RNAs (LncRNAs) have been linked in a wide range of biological processes in recent years, with the introduction of microarray and high-throughput sequencing technologies, and previous studies have established their tight relationship with several stages of NAFLD development. Existing studies have shown that lncRNAs can regulate the signaling pathways related to hepatic lipid metabolism, NASH, NASH-related fibrosis and HCC. This review aims to provide a basic overview of NAFLD and lncRNAs, summarize and describe the mechanisms of lncRNAs action involved in the development of NAFLD, and provide an outlook on the future of lncRNAs-based therapy for NAFLD.
La enfermedad del hígado graso no alcohólico (NAFLD) es la enfermedad hepática más común en el mundo, con estudios epidemiológicos que indican una prevalencia del 25%. La NAFLD se considera una enfermedad progresiva que avanza de esteatosis hepática simple a esteatohepatitis no alcohólica (NASH), luego a fibrosis hepática y, finalmente, a cirrosis o carcinoma hepatocelular (HCC). La investigación existente ha dilucidado principalmente la etiología de NAFLD. Sin embargo, sus procesos moleculares particulares siguen siendo inciertos. Los ARN largos no codificantes (lncRNA) se han relacionado en una amplia gama de procesos biológicos en los últimos años, con la introducción de microarrays y tecnologías de secuenciación de alto rendimiento, y estudios previos han establecido su estrecha relación con varias etapas del desarrollo de NAFLD. Los estudios existentes han demostrado que los lncRNA pueden regular las vías de señalización relacionadas con el metabolismo lipídico hepático, NASH, fibrosis relacionada con NASH y HCC. Esta revisión tiene como objetivo proporcionar una visión general básica de NAFLD y lncRNA, resumir y describir los mecanismos de acción de lncRNA involucrados en el desarrollo de NAFLD, y proporcionar una perspectiva sobre el futuro de la terapia basada en lncRNA para NAFLD.
Non-alcoholic fatty liver disease (NAFLD) is a chronic liver disease in which the fatty degeneration of the liver reaches 5–10% or more of the total liver weight. However, the disease is not due to excessive alcohol consumption (≤20g/d for women and ≤30g/d for men) or other causes.1 The prevalence of NAFLD in the general population is as high as 25%.2 Current research suggests that NAFLD is close association with obesity and type 2 diabetes mellitus (T2DM).3 It is estimated that 47.3–63.7% of patients with type 2 diabetes4 and up to 80% of obese patients5 develop symptoms related to NAFLD. NAFLD can progress from simple steatosis to non-alcoholic steatohepatitis (NASH). If not effectively controlled, it may eventually progress to cirrhosis or even hepatocellular carcinoma (HCC), seriously affecting physical health.6 As scientists have studied NAFLD, they have found that the main risk factors associated with NAFLD progression include age, inflammation, intestinal-liver axis signaling, genetic polymorphisms such as single nucleotide polymorphisms in the PNPLA3 gene1 and metabolic abnormalities such as obesity, insulin resistance, and type 2 diabetes. In addition, diet can have a significant impact on NAFLD. In addition to the well-known high-fat diet, fructose intake has the potential to induce hepatic lipid deposition and hepatic steatosis in humans.7
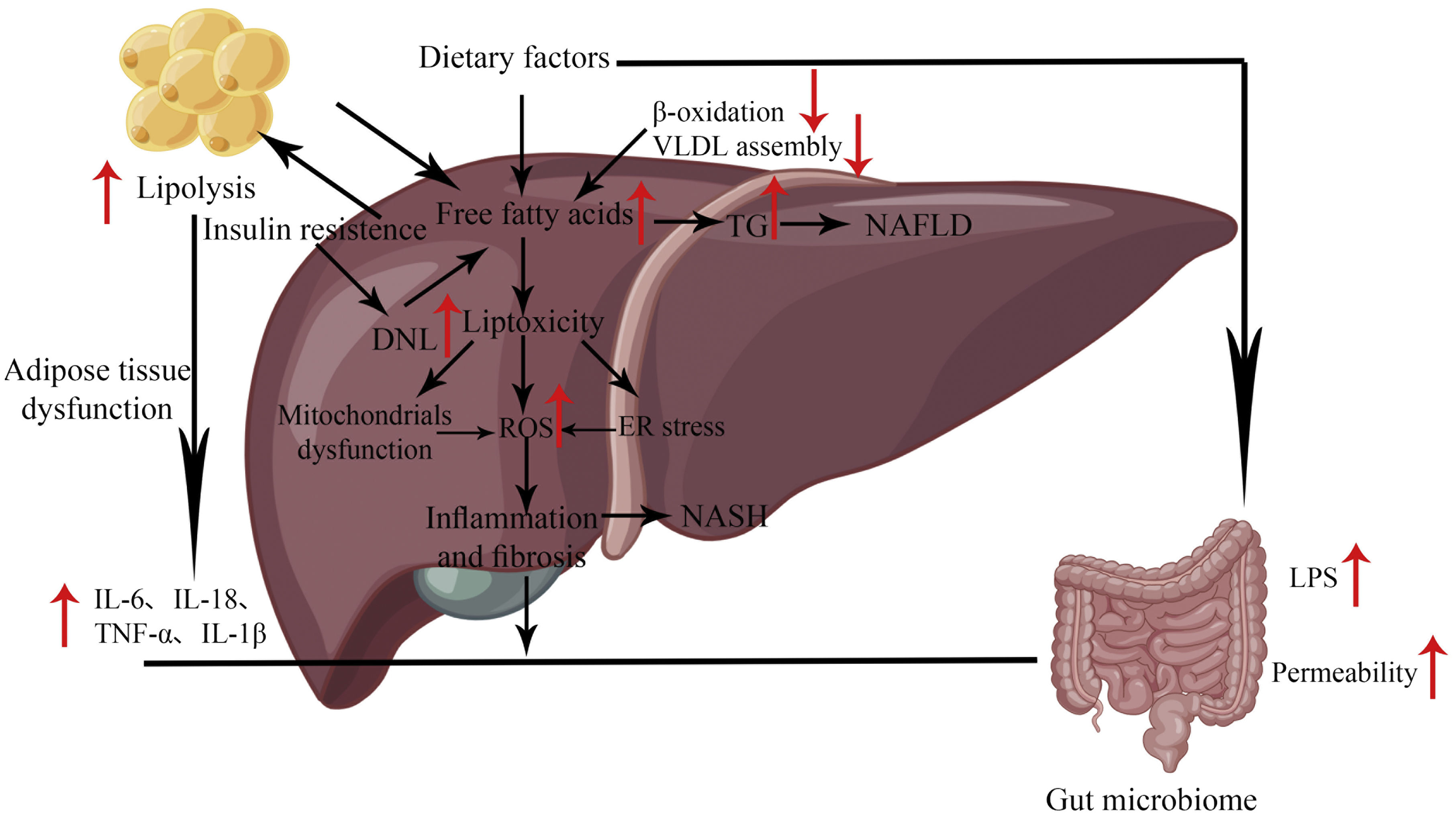
Because of the complexity of the specific pathogenesis of NAFLD (Fig. 1), no definitive conclusions can be drawn about its pathogenesis. The “two-hit theory” is a classic theory that elucidates the pathogenesis of NAFLD. The theory states that NAFLD develops after two hits. The first being the excessive accumulation of fat in the liver, which subsequently triggers a second hit (cytotoxic event), including the release of inflammation-related cytokines (e.g.TNFα, IL-1β, IL-6,etc.) and adipokines, mitochondrial dysfunction, and oxidative stress, leading to hepatocyte damage.8 However, this theory is somewhat flawed in failing to address the molecular and metabolic mechanisms behind the pathogenesis of steatosis and NASH. It does not fully explain the development of lipid deposition in hepatocytes during the development of NAFLD.

In some cases, inflammation may precede steatosis, so it is wrong to think of NAFLD as a disease continuum. In addition, simple steatosis can have a protective effect on the liver.9 For these defects, scientists later developed the “multiple hit theory,” which states that excessive accumulation of liver fat remains the first hit but that hepatocytes are affected to varying degrees by genetic and epigenetic factors, changes in the gut microbiota and endoplasmic reticulum stress (ERS), resulting in widespread metabolic dysfunction and accelerated disease progression.9
Long non-coding RNAs (LncRNAs) are a group of molecules longer than 200 nucleotides that are not generally involved in protein-coding and are therefore often referred to by scholars as ‘noisy sequences’.10LncRNAs can be classified into five categories depending on their position in the genome: antisense lncRNAs, intergenic lncRNAs, intronic lncRNAs, bidirectional lncRNAs, and sense lncRNAs. The lncRNAs has the exact transcription from corresponding genes as the mRNAs, and after shearing has 5′ cap and 3′ polyA tail that can turn the same gene into a different transcript by variable shearing.11 The difference is that it does not have the ability of mRNAs to encode proteins, and it is differently located, with different lncRNAs differing in abundance inside (and outside) the cell.
Studies have shown that the influence mechanisms of lncRNAs mainly include the following: (a) Guiding target localization; (b) Acting as a molecular scaffold mediating protein-RNA interactions; (c) Act as a miRNA sponge; (d) Act as a molecular decoy to bind directly to proteins and inhibit downstream gene expression; (e) Encode transcription of upstream promoters of target genes and interfere with chromatin remodeling and histone modification to affect downstream gene expression; (f) As a precursor of small RNA. In addition, studies have also confirmed that lncRNAs can interact directly with signaling receptors.12 Scientists have discovered that lncRNAs can be involved in a wide range of biological activities, including epigenetic regulation, regulation of protein complexes, chromosome recruitment as well as inactivation, and regulation of cell growth and apoptosis, etc. For example, the lncRNADANCR can promote migration, invasion, and relocation of pre-eclamptic trophoblast cells via miR-214-5p.13 The abnormal functional expression of lncRNAs has also been reported to be closely associated with developing various diseases, such as cancer, inflammation, and liver disease. The lncRNA HSD11B1-AS1 is upregulated in the acute phase of Kawasaki disease, and interaction between HSD11B1-AS1 and G0S2 can occur to control inflammation in the acute phase of Kawasaki disease.14
A variety of dysregulated lncRNAs have been reported in NAFLD, which can play a crucial role in lipid accumulation, NASH, NASH-related fibrosis, and HCC through a variety of mechanisms, such as acting as competitive endogenous RNAs (ceRNAs), binding to RNA-binding proteins and controlling their phosphorylation, acetylation, and ubiquitination at the post-translational level. This review aims to examine the role of lncRNAs in the development of NAFLD and its future therapeutic prospects, further illuminating the future direction of NAFLD.
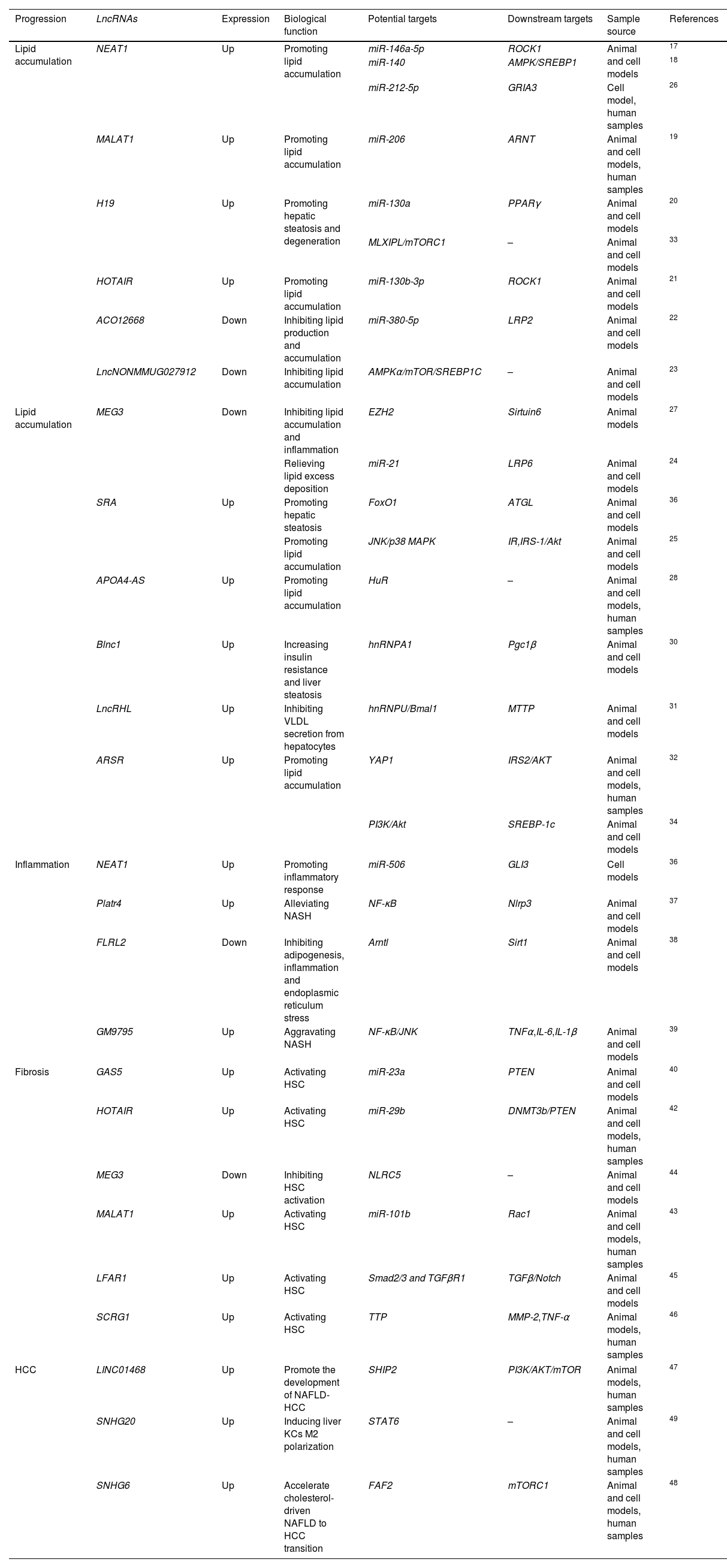
The role of lncRNAs in the development of NAFLDTo date, scientists have identified many abnormal lncRNAs expressions in NAFLD. Studies have shown that 1735 lncRNAs and 1485 mRNAs are differentially expressed in NAFLD samples compared to standard liver samples.15 They are widely involved in hepatic lipid metabolism, NASH, NASH-associated fibrosis and NAFLD-related HCC (Table 1). This chapter will outline the mechanisms of lncRNAs action in NAFLD (Fig. 2).
The mechanism of lncRNAs in NAFLD.
| Progression | LncRNAs | Expression | Biological function | Potential targets | Downstream targets | Sample source | References |
|---|---|---|---|---|---|---|---|
| Lipid accumulation | NEAT1 | Up | Promoting lipid accumulation | miR-146a-5p | ROCK1 | Animal and cell models | 17 |
| miR-140 | AMPK/SREBP1 | 18 | |||||
| miR-212-5p | GRIA3 | Cell model, human samples | 26 | ||||
| MALAT1 | Up | Promoting lipid accumulation | miR-206 | ARNT | Animal and cell models, human samples | 19 | |
| H19 | Up | Promoting hepatic steatosis and degeneration | miR-130a | PPARγ | Animal and cell models | 20 | |
| MLXIPL/mTORC1 | – | Animal and cell models | 33 | ||||
| HOTAIR | Up | Promoting lipid accumulation | miR-130b-3p | ROCK1 | Animal and cell models | 21 | |
| ACO12668 | Down | Inhibiting lipid production and accumulation | miR-380-5p | LRP2 | Animal and cell models | 22 | |
| LncNONMMUG027912 | Down | Inhibiting lipid accumulation | AMPKα/mTOR/SREBP1C | – | Animal and cell models | 23 | |
| Lipid accumulation | MEG3 | Down | Inhibiting lipid accumulation and inflammation | EZH2 | Sirtuin6 | Animal models | 27 |
| Relieving lipid excess deposition | miR-21 | LRP6 | Animal and cell models | 24 | |||
| SRA | Up | Promoting hepatic steatosis | FoxO1 | ATGL | Animal and cell models | 36 | |
| Promoting lipid accumulation | JNK/p38 MAPK | IR,IRS-1/Akt | Animal and cell models | 25 | |||
| APOA4-AS | Up | Promoting lipid accumulation | HuR | – | Animal and cell models, human samples | 28 | |
| Blnc1 | Up | Increasing insulin resistance and liver steatosis | hnRNPA1 | Pgc1β | Animal and cell models | 30 | |
| LncRHL | Up | Inhibiting VLDL secretion from hepatocytes | hnRNPU/Bmal1 | MTTP | Animal and cell models | 31 | |
| ARSR | Up | Promoting lipid accumulation | YAP1 | IRS2/AKT | Animal and cell models, human samples | 32 | |
| PI3K/Akt | SREBP-1c | Animal and cell models | 34 | ||||
| Inflammation | NEAT1 | Up | Promoting inflammatory response | miR-506 | GLI3 | Cell models | 36 |
| Platr4 | Up | Alleviating NASH | NF-κB | Nlrp3 | Animal and cell models | 37 | |
| FLRL2 | Down | Inhibiting adipogenesis, inflammation and endoplasmic reticulum stress | Arntl | Sirt1 | Animal and cell models | 38 | |
| GM9795 | Up | Aggravating NASH | NF-κB/JNK | TNFα,IL-6,IL-1β | Animal and cell models | 39 | |
| Fibrosis | GAS5 | Up | Activating HSC | miR-23a | PTEN | Animal and cell models | 40 |
| HOTAIR | Up | Activating HSC | miR-29b | DNMT3b/PTEN | Animal and cell models, human samples | 42 | |
| MEG3 | Down | Inhibiting HSC activation | NLRC5 | – | Animal and cell models | 44 | |
| MALAT1 | Up | Activating HSC | miR-101b | Rac1 | Animal and cell models, human samples | 43 | |
| LFAR1 | Up | Activating HSC | Smad2/3 and TGFβR1 | TGFβ/Notch | Animal and cell models | 45 | |
| SCRG1 | Up | Activating HSC | TTP | MMP-2,TNF-α | Animal models, human samples | 46 | |
| HCC | LINC01468 | Up | Promote the development of NAFLD-HCC | SHIP2 | PI3K/AKT/mTOR | Animal models, human samples | 47 |
| SNHG20 | Up | Inducing liver KCs M2 polarization | STAT6 | – | Animal and cell models, human samples | 49 | |
| SNHG6 | Up | Accelerate cholesterol-driven NAFLD to HCC transition | FAF2 | mTORC1 | Animal and cell models, human samples | 48 | |
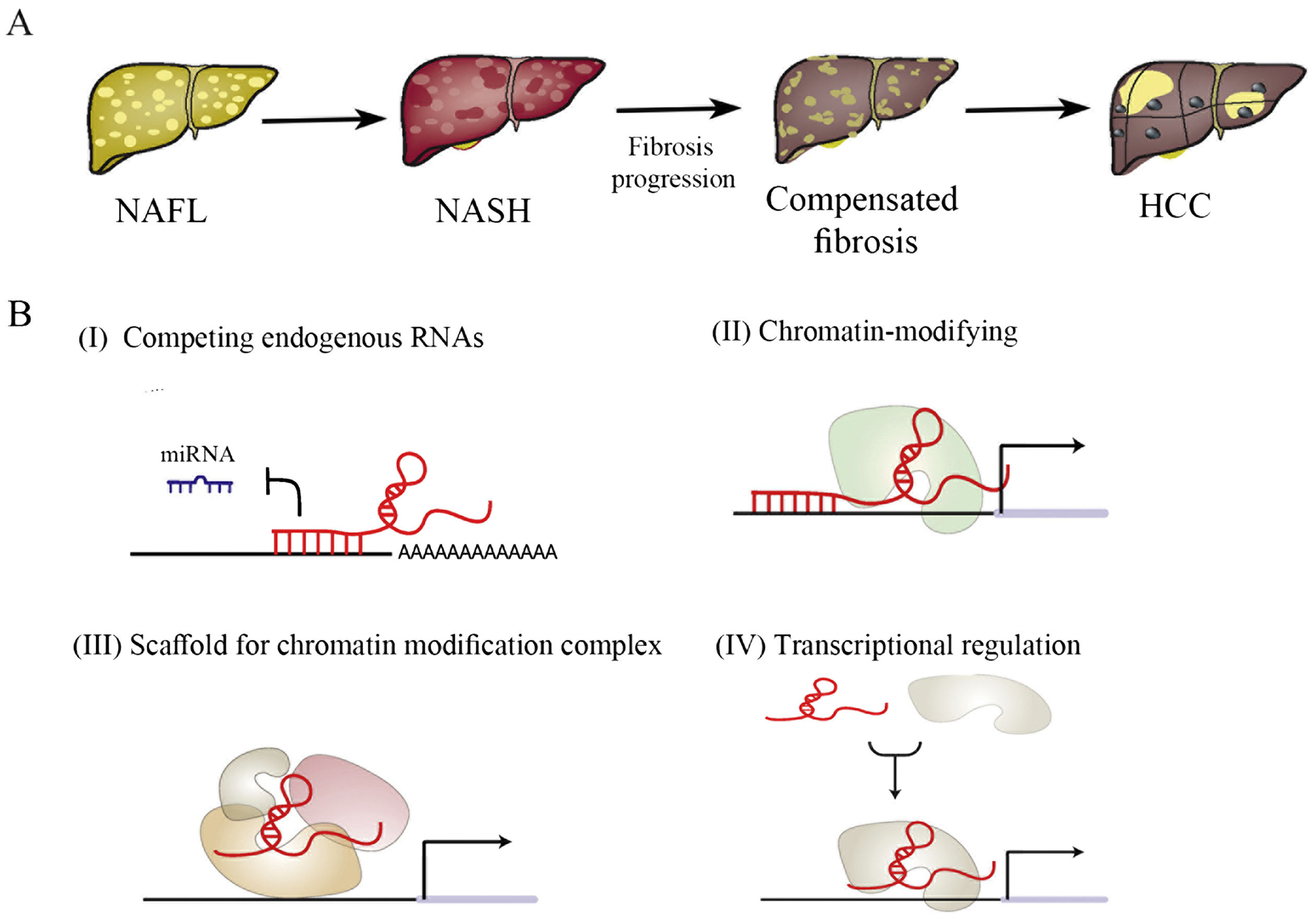
 Progression of NAFLD. (B) The mechanism of lncRNAs in NAFLD: (I) LncRNAs function as molecular sponges and compete for miRNA binding to mRNAs, thus inhibiting miRNA-mediated gene repression; (II) LncRNAs act as guide by recruiting chromatin-modifying enzymes to target genes, either in cis or in trans to distant target genes; (III) LncRNAs bring together multiple proteins to form ribonucleoprotein complexes, thus acting as a scaffold; (IV) LncRNAs interact directly with transcription factors to activate/inhibit the expression of target genes. The pathogenesis of NAFL includes the above four mechanisms. The pathogenesis of NASH is mainly mechanism (I) and (IV), while the relevant mechanism of NASH-related fibrosis is (I) and (II). The pathogenesis of NAFLD-HCC is mainly mechanism (II) and (III).")
(A) Progression of NAFLD. (B) The mechanism of lncRNAs in NAFLD: (I) LncRNAs function as molecular sponges and compete for miRNA binding to mRNAs, thus inhibiting miRNA-mediated gene repression; (II) LncRNAs act as guide by recruiting chromatin-modifying enzymes to target genes, either in cis or in trans to distant target genes; (III) LncRNAs bring together multiple proteins to form ribonucleoprotein complexes, thus acting as a scaffold; (IV) LncRNAs interact directly with transcription factors to activate/inhibit the expression of target genes. The pathogenesis of NAFL includes the above four mechanisms. The pathogenesis of NASH is mainly mechanism (I) and (IV), while the relevant mechanism of NASH-related fibrosis is (I) and (II). The pathogenesis of NAFLD-HCC is mainly mechanism (II) and (III).
The early stage of NAFLD is hepatic steatosis, mainly characterized by excessive triglyceride (TG) deposition in the hepatocytes. This is mainly caused by an imbalance between TG/fatty acid acquisition and removal, which involves an increase in the uptake of circulating lipids and de novo lipogenesis (DNL), as well as a decrease in fatty acid oxidation (FAO) and lipid output from very low-density lipoproteins (VLDL). DNL is an essential source of TG deposition in the liver, and the transcription factors ChREBP and SREBP-1c play a vital role in this process. Their activation promotes the expression of downstream lipogenic genes (FAS, ACC1, etc.), thereby stimulating DNL. In addition, hepatic TG utilization is primarily β-oxidation of fatty acids, with PPARα/PGC-1α playing a crucial role. PPARα is activated by fatty acids, and upon their binding, it can promote the expression of fatty acid metabolism precursor genes.
NAFLD has been treated as a metabolic disease, which has led to impaired lipid metabolism being seen as key to the pathogenesis of NAFLD. The role of lncRNAs in regulating hepatic lipid metabolism has been widely studied. lncRNAs can protect their protein-coding counterparts from post-translational regulation by isolating miRNAs. This ceRNA mechanism has been adopted as the primary molecular mechanism regulating lipid metabolism in NAFLD. For example, lncRNA uc.372 was reported to reduce the expression inhibition of target genes related to lipid synthesis and uptake (ACC, FAS, SCD1, CD36) by binding to pri-miR-195/pri-miR-4668 and inhibiting the maturation of miR-195/miR-4668.16 This leads to lipid accumulation in the liver and aggravates NAFLD. In addition, AMPK is an essential regulator of lipid and glucose metabolism, and activation of AMPK inhibits SREBP1 expression. Chen et al. showed that lncRNA NEAT1 could affect lipid accumulation in NAFLD by targeting miR-146a-5p, resulting in reduced expression of miR-146a-5p and increased expression of the downstream target ROCK1, which in turn affects the AMPK/SREBP pathway.17 Sun et al. found another mechanism that NEAT1 could also directly bind to miR-140 and exert a synergistic effect with miR-140 to inhibit AMPK/SREBP1 signaling, thereby increasing lipid accumulation and aggravating NAFLD.18 In NAFLD, lncRNA MALAT1 can upregulate the expression of its target ARNT by binding to miR-206, which can bind to the PPARα promoter, thereby inhibiting PPARα expression and inducing upregulation of CD36 expression, leading to hepatic fat accumulation.19 In addition, PPARγ is another crucial transcription factor in liver metabolism and is highly expressed in fat. Liu et al. demonstrated that lncRNA-H19 could bind to miR-130a and promote PPARγ expression through downregulating miR-130a expression levels, which promoted hepatic lipogenesis and degeneration and the expression of NAFLD-related genes.20 That high expression of GW9662 (an antagonist of miR-130a and PPARγ) inhibited hepatic lipogenesis.20 In addition, other lncRNAs are present in this mechanism, including HOTAIR,21ACO12668,22LncNONMMUG027912,23 and MEG3.24
There are other molecular mechanisms for lncRNAs in this process. Some lncRNAs can also regulate protein degradation or production by interacting with histone-modifying enzymes to control their phosphorylation, acetylation, and ubiquitination, which affect their target genes expression levels. This mechanism has also proved to be expected in lipid metabolism. For example, lncRNA SRA can promote adipogenesis by inhibiting JNK/p38 MAPK, increasing insulin receptor (IR) gene expression, and increasing phosphorylation of the downstream target IRS-1/Akt.25H3K27 is present at the promoter of NEAT1 and is enriched here. H3K27 acetylation promotes the transcriptional level of lncRNA NEAT1. Upregulation of NEAT1 targets miR-212-5p, decreases miR-212-5p expression levels, and subsequently upregulates GRIA3 expression, promoting hepatic lipid accumulation.26 Overexpression of lncRNA MEG3 can inhibit the development of NAFLD by promoting the ubiquitination and degradation of EZH2, downregulating the expression of EZH2, and upregulating the expression of Sirtuin6 (a target gene of EZH2), which exerts inhibition of lipid accumulation and inflammation.27
Several studies have shown that the mechanism that lncRNAs can act as a central platform to bind to different proteins and facilitate their intermolecular interactions also plays an essential regulatory role in hepatic lipid metabolism. It has been reported that lncRNAs can bind to RNA-binding proteins to take effect. For example, lncRNA ApoA4-AS is co-expressed with APOA4, and ApoA4-AS can physically interact with HuR (mRNA stabilizing protein) to form a complex with APOA4mRNA to stabilize APOA4mRNA and increase the expression of APOA4-AS and APOA4.28 This increased plasma TG and total cholesterol levels in ob/ob mice and aggravated lipid accumulation in the liver.29 Another recent study showed that HuR can also inhibit liver lipid accumulation by inhibiting the expression of lncRNA H19 and reducing SphK2 nuclear translocation.29LncRNA Blnc1 can bind to hnRNPA1 to increase the expression of Pgc1β, thereby aggravating insulin resistance and hepatic steatosis.30LncRHL can directly bind to hnRNPU to make it more stable and avoid degradation, activating the expression of Bmal1 and reducing the expression of its downstream target MTTP, thus inhibiting VLDL secretion in hepatocytes.31 In addition, lncRNAs can also bind to other proteins and play a role. For example, lncRNA ARSR can specifically bind to YAP1, promote the nuclear translocation of YAP1 and inhibit the phosphorylation of YAP1, thereby activating the IRS2/AKT pathway and further increasing lipid accumulation in vivo and in vitro.32
Recent studies have also revealed that lncRNAs can activate or inhibit the expression of genes involved in pathways related to hepatic lipid metabolism by regulating the interaction of cellular signaling pathways and transcription factors. For example, lncRNA-H19 can induce hepatic steatosis by activating the MLXIPL and mTORC1 networks in hepatocytes.33 The lncRNAARSR can regulate hepatic fat accumulation by increasing SREBP-1c expression by activating the PI3K/Akt pathway.34LncRNASRA can promote steatosis by inhibiting the transcriptional activity of FoxO1, reducing the expression of its downstream target ATGL and then decreasing free fatty acid (FFA) β-oxidation in hepatocytes.35
In summary, various mechanisms extensively involve lncRNAs in hepatic lipid metabolism. This indicates the need for scientists to investigate the mechanisms of lncRNAs in lipid metabolism. In the future, we need to continue to conduct further mechanistic studies on the aberrantly expressed lncRNAs, to investigate further the deeper mechanisms of lncRNAs in the development of NAFLD.
The role of lncRNAs in NASHNAFLD stimulates NASH by sustaining lipid degeneration in the liver and promoting the formation of toxic lipid metabolites. Available studies also confirm that NASH is the next step in developing NAFLD. Pro-inflammatory factors play a vital role in this development. After years of research, scientists found that lncRNAs are potential regulators in NASH that can mediate inflammation by enhancing endoplasmic reticulum stress or directly activating inflammation-related signaling pathways.
In NASH, lncRNAs act by a molecular mechanism similar to lncRNA-mediated fat accumulation. It can act as ceRNAs to mediate the production of inflammation in hepatocytes. For example, it has been confirmed that lncRNA NEAT1 is highly expressed and miR-506 is lowly expressed in NAFLD progression, and they have also been shown to regulate inflammatory response. Further mechanistic studies showed that miR-506 could bind to NEAT1 and GLI3, and NEAT1 could sponge miR-506 to regulate GLI3 expression. This suggests that NEAT1 regulates the inflammatory response of NASH through the miR-506/GLI3 axis.36
Several studies have shown that lncRNAs mediate NASH production through other mechanisms. It is reported that lncRNAs can be regulated through the activation and inflammatory signaling pathways that play the role of NASH. For example, lncRNAPlatr4 improves NASH by preventing NF-κB/Rxrα complexes from binding to κB sites through physical interactions, thereby blocking the NF-κB signaling pathway to inactivate Nlrp3 inflammatory vesicles.37LncRNA FLRL2 inhibits adipogenesis, inflammation, and ERS by activating the Arntl-Sirt1 axis.38LncRNAGM9795 is highly expressed in the liver tissue of the NASH animal and NASH cell models. It can enhance ERS and activate NF-κB/JNK signaling pathway, thereby affecting the expression of inflammatory factors (TNFα, IL-6, IL-1β) and promoting the development of NASH.39
The above shows that lncRNAs is also essential in the inflammatory response of NASH, but the mechanism of this aspect still needs to be better studied as lipid metabolism. In the future, we need to study more inflammation-related pathways to find new mechanisms and provide new possibilities for treating NASH. In addition, lncRNAs can regulate the inflammatory response of NASH patients, which may become a potential non-invasive biomarker to provide more convenience for clinical diagnosis and treatment.
The role of lncRNAs in NASH-associated fibrosisFibrosis results from the further development of NASH and is a severe stage of the development of NAFLD. Hepatic stellate cells (HSC) are the key cells that influence the development of liver fibrosis. In the ordinary course of events, the HSC is stable. However, when the HSC is stimulated by inflammation, pathological accumulation of extracellular matrix (ECM) components in the liver can occur, leading to fibrosis. Through years of research, new pathways and mediators mediating the development of fibrosis have been identified, not least endoplasmic reticulum stress, oxidative stress, epigenetics, and receptor-mediated signaling. This suggests that the mechanisms of HSC activation are complex. Existing studies confirm that many types of lncRNAs can play a crucial role in the activation, proliferation, or apoptosis of HSC, which can further influence the progression of liver fibrosis.
The most common of the multiple mechanisms by which lncRNAs mediate NASH-associated fibrosis is their involvement in regulating liver fibrosis as ceRNAs that bind to miRNAs. For example, the lncRNA GAS5 can act as a sponge for miR-23a, competitively reducing the expression of miR-23a, which can target PTEN, thereby activating the downstream signaling molecule PI3K - p85/Akt/mTOR pathway, resulting in decreased levels of the epithelial marker E-cadherin and increased levels of the myofibroblast marker α-SMA and the extracellular matrix protein collagen I, ultimately leading to liver fibrosis and HSC activation.40 Another study confirmed that GAS5 is not only associated with NASH-related fibrosis but also closely related to cirrhosis.41 This could perhaps be another direction for our future research. In addition, lncRNAHOTAIR can competitively bind to miR-29b, reducing the expression of miR-29b, restoring its target DNMT3b, and enhancing PTEN methylation, thereby reducing PTEN and increasing the expression of collagen I and α-SMA, leading to HSC activation and promoting the development of liver fibrosis. Knockdown of HOTAIR can inhibit liver fibrosis by restoring miR-29b and inhibiting DNMT3b, reduce PTEN methylation and increase PTEN level, thereby inhibiting liver fibrosis.42 The lncRNAMALAT1 can act as a ceRNA for miR-101b, reducing the expression of miR-101b and increasing the expression of its target Rac1, thereby promoting HSC proliferation and activation.43
It has been reported that another common mechanism is that lncRNAs can directly bind to proteins to exert regulatory effects to mediate the production of NASH-associated fibrosis. The lncRNAMEG3 inhibits HSC activation and reverses liver fibrosis by targeting the NLRC5 protein.44LncRNALFAR1 can promote the binding between Smad2/3 and TGFβR1, and promote the expression and phosphorylation of Smad2/3. It can also directly bind to Smad2/3 to regulate the transcription of TGFβ, Smad2, Smad3, Notch2, Notch3 and Hes1, activate TGFβ/Notch pathway to activate HSC and promote the progression of liver fibrosis.45 The lncRNA SCRG1 can bind specifically to the RNA-binding protein TTP. Its overexpression can lead to TTP mRNA instability, reducing TTP expression and decreasing TTP expression the expression of its targets MMP-2 and TNF-α and promotes HSC activation and liver fibrosis progression.46
In conclusion, lncRNAs plays a vital role in NASH-related fibrosis and also illustrates that HSC activation is central to liver fibrosis. Existing studies have focused on the genes and signaling pathways associated with HSC activation. In the future, we could further research to explore other mechanisms of lncRNAs involvement in fibrosis. Furthermore, these studies suggest that lncRNAs have great potential in NAFLD research and new approaches to treat NASH-related fibrosis could be further explored.
Role of lncRNAs in NAFLD-related HCCNAFLD-HCC is a complex and incompletely studied process. During this process, various pathophysiological changes such as insulin resistance, cytokine release, oxidative stress and mitochondrial damage may occur. However, hepatic lipid storage, the most important characteristic of NAFLD, is a key factor in promoting HCC development. FFA and diglycerol can facilitate ERS and reactive oxygen-species-mediated DNA damage, which leads to a chronically inflamed hepatic environment, resulting in NASH and liver fibrosis, which will further drive the oncogenesis of NAFLD-related HCC. NASH is key to this process. Studies have shown that HOTAIR and MALAT1 are highly expressed in NASH and are associated with tumor cell proliferation, migration and invasion. This suggests that they may be important markers of NASH and NASH-HCC. However, more in-depth studies are still needed to reveal the specific expression of lncRNAs and their mechanism of action in NASH-HCC.
Most current studies focus on NAFLD-HCC. Recent studies have shown that LINC01468 can activate the PI3K/AKT/mTOR signaling pathway by binding SHIP2 and promoting the ubiquitination and degradation of CUL4A, thereby promoting lipogenesis and accelerating the development of HCC.47 Previous studies have confirmed that hepatic cholesterol is one of the main lipotoxic molecules involved in the progression of NAFLD to HCC. However, lncRNA SNHG6 is involved in this process. Because SNHG6 is located at the endoplasmic reticulum – lysosome contact site, it can regulate cholesterol-dependent mTORC1 lysosomal recruitment and activation, promote cholesterol synthesis, and accelerate NAFLD-HCC.48 These studies indicate that it is necessary to investigate the role of lncRNAs in NAFLD-HCC, but there are still few studies on this topic. Furthermore, previous suggests that polarization of liver macrophage Kupffer cells (KCs) may be a contributing factor to NAFLD-associated HCC. Wang et al. found that lncRNA SNHG20 expression was decreased in human NAFLD, but increased in human NAFLD-HCC liver. SNHG20 may facilitate the progression of NALFD to HCC via inducing liver KCs M2 polarization via STAT6 activation.49
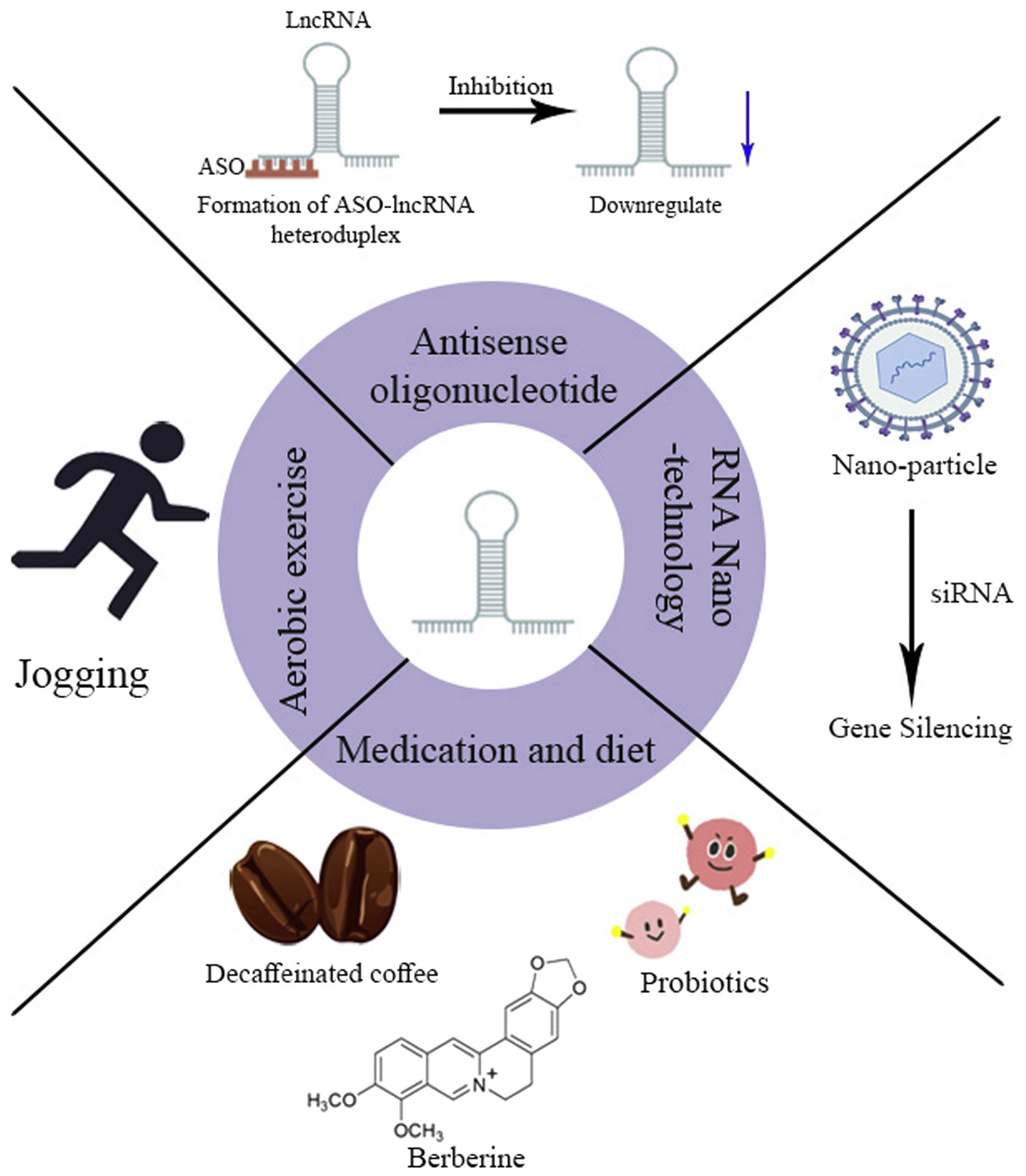
Prospects for lncRNAs-based therapy for NAFLDBecause of the crucial role of lncRNAs in the development of NAFLD and the presence of multiple lncRNAs aberrantly expressed in NAFLD. This offers the possibility of lncRNAs-based therapies to correct this dysregulation. Currently, NAFLD is treated mainly through lifestyle changes. It has been reported that dietary intervention based on Mediterranean diet and lifestyle changes is the main means of treatment for NAFLD.51 For example, aerobic exercise may inhibit the transcriptional activity of FoxO1 by suppressing the expression of lncRNASRA, which leads to upregulation of ATGL expression, reducing intrahepatic lipid accumulation, and inhibiting inflammatory proteins and the JNK/p38MAPK signaling pathway, allowing for further relief of inflammation.52 However, compliance with this treatment is relatively poor and the therapeutic effect is difficult to maintain. Therefore, we need to develop new treatment strategies for NAFLD (Fig. 3).
 can regulate the expression of lncRNAs to alleviate NAFLD. In addition, oligonucleotide and RNA nanotechnology can also achieve therapeutic goals by targeting silencing lncRNAs.")
In recent years, numerous achievements in studying the mechanisms of lncRNAs action in NAFLD have accelerated the research on lncRNAs-based therapies for NAFLD. Decaffeinated coffee has been reported to restore lncRNAGM16551 to normal levels and to downregulate the expression of its target genes ACC1 and SCD1. In addition, it significantly reduces the expression of lncRNAH19 and its target gene collagen α-1(I) chain and normalizes the expression level of αSMA protein, thereby preventing NAFLD.53 Studies have confirmed that the biologically active substance flavopiridol (BBR) in Chinese medicine can be used to treat a various diseases.54 BBR may treat NAFLD by upregulating the expression of the lncRNA MRAK052686 and its associated gene Nrf2.55 In addition, the probiotic mixture, which can be used alone or combined with prebiotic inulin fiber, can improve hepatic steatosis, inflammation, and fibrosis by modulating a panel of Hippo signaling pathway-associated RNAs, downregulating the expression of YAP1 and miR-1205, and regulating the expression of LATS1, NF2, and the lncRNA SRD5A3-AS1.56 These studies reveal that these exogenous materials have the potential to alter lncRNAs expression, offering a new therapeutic approach for the treatment of NAFLD. However, research into its corresponding mechanisms should be deepened in the future.
Gene-targeted therapyAntisense oligonucleotides (ASOs) are chemically synthesized nucleic acid analogs, usually around 12–30 nucleotides in length, which can regulate RNAs processing and protein expression through different mechanisms (promoting RNAs cleavage and degradation or occupancy-only mechanisms) and can act as targets for a variety of molecules. With advanced research into the structure and chemistry of ASOs, they are already being used as a valuable tool for the future treatment of many diseases. Many studies have confirmed the therapeutic role of ASOs in the disease. For example, LINC00680 promotes proliferation, migration, and invasion of esophageal squamous cell carcinoma (ESCC) and is strongly associated with poor patient prognosis. Xue et al. designed specific targeting ASO LINC00680 and negative form control ASO NC, transfected with ASOs into KYSE510 and KYSE140 cells, following injection of ASO LINC00680-targeted treatment in a mouse model of hetero-transplanted KYSE510 cells, they found that tumor growth was significantly inhibited in the ASO LINC00680-treated group compared to the ASO NC-treated group and that the expression levels of LINC00680 were significantly reduced. This suggests that ASOs targeting LINC00680 may be able to suppress it expression, with the promise of lncRNAs-based therapy for ESCC.57 From the above description, we can draw a welcome conclusion that ASO can be used for lncRNAs function loss. It indicates that we can use the existing theory to design ASOs-targeted lncRNAs-based therapies for treating NAFLD.
In recent years, RNA nanotechnology has flourished and has been adopted as a novel delivery system for targeted therapies for various human diseases. The lncRNATMEM1-AS5 has been reported to be key in regulating GC drug resistance. Zhou et al. constructed a novel chitosan-gelatin-EGCG (CGE) nanocarrier for selective delivery of si-TMEM44-AS1 to silence TMEM44-AS1 expression and exert a reversal of 5-FU resistance in GC, thereby enhancing 5-FU efficacy in a GC xenograft nude mouse model.58 In conclusion, nanoparticles can treat relevant diseases by selectively delivering siRNAs, thereby silencing the relevant disease-causing genes. These results suggest that this therapeutic approach based on RNA nanoparticle-mediated lncRNAs expression holds excellent promise. In the future, we can prepare RNA nanoparticles to silence the expression of NAFLD-related lncRNAs to treat NAFLD, which requires our future in-depth research.
Conclusion and outlookIn conclusion, many lncRNAs are expressed differently in NAFLD and are involved in multiple processes in the development of NAFLD. In this review, we summarize the mechanisms of lncRNAs involvement in hepatic lipid metabolism, NASH, NASH-associated fibrosis, and NAFLD-related HCC. In addition, we look forward to the future potential of lncRNAs in the treatment of NAFLD to be developed into a clinical lncRNAs-based treatment of NAFLD. For example, coffee, flavonoid, ASOs, and RNA nanotechnology are used. It has been shown that lncRNAs play a key role in diagnosing and treating NAFLD. Future research is needed to continue investigating the regulatory mechanisms of lncRNAs in NAFLD, further revealing their potential role as diagnostic and therapeutic targets and prognostic biomarkers of NAFLD.
Conflict of interestThe authors declare that they have no conflict of interest.

