




El síndrome de Alagille (SA) (SALG; OMIM 118450) tiene una incidencia aproximada de 1/30.000 recién nacidos vivos1. Es una enfermedad multisistémica con patrón de herencia autosómico dominante, con penetrancia prácticamente completa, aunque aproximadamente un 50-70% de los casos son esporádicos, producidos por mutaciones de novo y, además, existe una gran variabilidad en su expresión clínica. Se trata de un síndrome caracterizado por manifestaciones hepáticas principalmente colestasis (96%), vértebras en mariposa (51%), embriotoxon posterior (78%), cardiopatía congénita, la más frecuente de ellas es la estenosis de la arteria pulmonar (97%) y facies peculiar (96%). El estudio anatomopatológico de la biopsia hepática muestra escasez de conductos biliares interlobulares. Se ha localizado el defecto genético en 2 genes diferentes. En más del 90% de los casos se han identificado mutaciones en el gen JAG11–3. El pronóstico depende fundamentalmente de la afectación hepática y las malformaciones cardiovasculares4,5. La evolución de la enfermedad es a una cierta mejoría espontánea en los primeros años de vida, aunque un 15-20% de los pacientes requerirán trasplante hepático5,6. En este artículo se describe un caso clínico de especial interés por la asociación de SA a atresia intestinal en el período neonatal, descrita en muy pocos casos en la bibliografía; y el diagnóstico del padre a raíz del diagnóstico del hijo con una expresividad mucho menos severa.
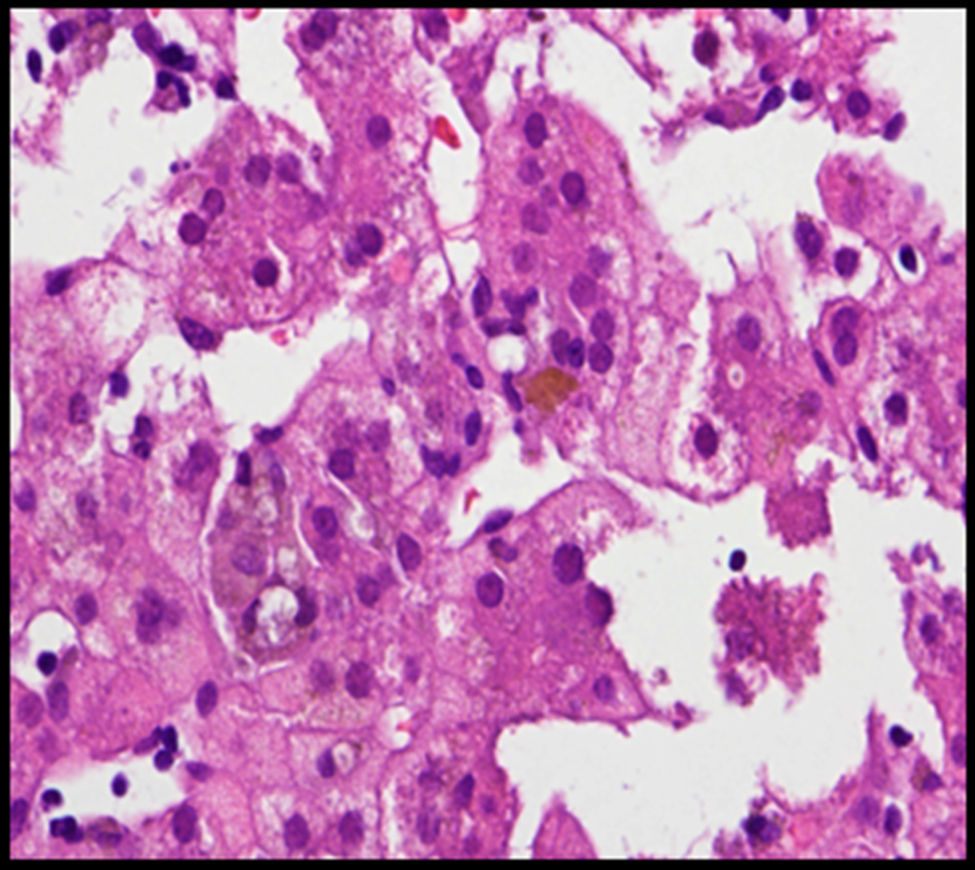
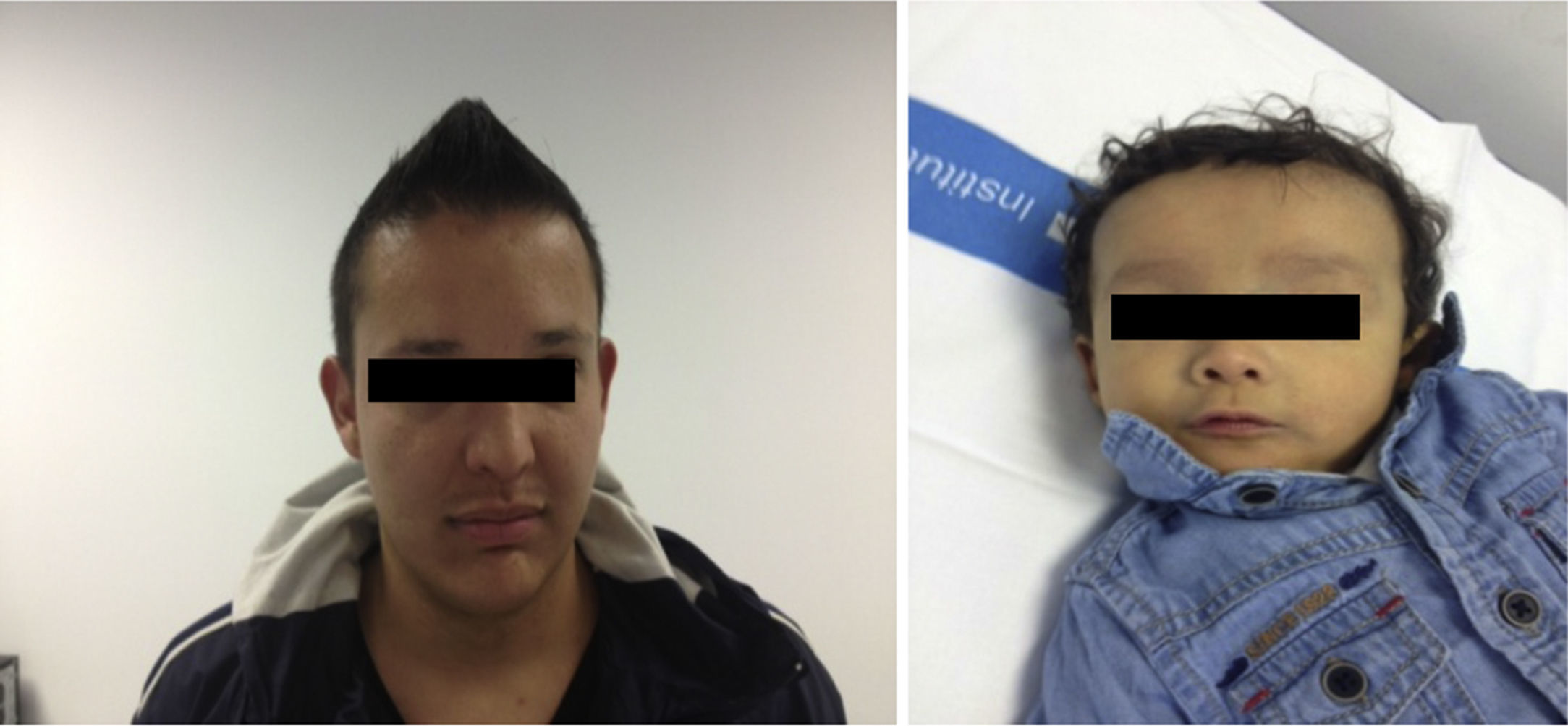
Se trata de un recién nacido a término, de peso adecuado, con estenosis de ramas pulmonares detectada por soplo cardíaco. Desde las primeras horas de vida destaca la dificultad para la alimentación, vómitos, distensión abdominal con ausencia de emisión de primer meconio. Se realiza radiografía de abdomen donde se observa ausencia de aire rectal y distensión de las asas a nivel del hemiabdomen superior, sospechándose de atresia intestinal, que se confirma. Se realiza intervención quirúrgica a las 48h de vida, practicándose resección de 23cm de intestino delgado proximales a la zona de la estenosis. Recibe nutrición parenteral durante 11 días. A partir del tercer día de vida presenta cuadro de ictericia no isoinmune con bilirrubina total máxima de 12,5mg/dl. A los 10 días de vida se observa patrón analítico de colestasis (bilirrubina total 6,41mg/dl, bilirrubina directa 4,75mg/dl, GGT 290U/l, AST 48U/l, ALT 26U/l), que se atribuye a nutrición parenteral, por lo que se suspende. Tras permanecer dicha colestasis en siguientes controles analíticos se realiza ecografía abdominal en la que se observa vesícula biliar con contenido en su interior, sin dilatación de vías biliares. Se aprecia empeoramiento progresivo de la colestasis hasta niveles máximos de bilirrubina directa de 10mg/dl, por lo que se amplía estudio etiológico. Se realiza estudio metabólico, hormonal, test del sudor, investigación de CMV y alfa-1-antitripsina, que resultan normales. En la colangio-RMN no se aprecian signos obstructivos extrahepáticos o de ramas principales hepáticas. Se realiza gammagrafía ácido iminodiacético hepatobiliar (HIDA), que muestra paso de contraste a duodeno. Se practica biopsia hepática en la que destaca paucidad de vías biliares (fig. 1) que, asociado a colestasis, cardiopatía y facies peculiar (fig. 2) sugiere el diagnóstico de SA. Se realiza estudio oftalmológico y radiografía vertebral que son normales. El patrón de colestasis sufre un empeoramiento progresivo (cifras máximas de GGT de 1.500U/l), con hiperbilirrubinemia conjugada alrededor de 12mg/dl a pesar del tratamiento con ácido ursodeoxicólico e hipercolesterolemia severa (máx 450mg/dl), apareciendo prurito de difícil control, junto con esplenomegalia progresiva secundaria a hipertensión portal, precisando trasplante hepático de donante vivo emparentado (madre) a los 2 años de edad sin incidencias y buena evolución posterior. El análisis del gen JAG1, detecta una transversión de C por A (c.756>A) en heterocigosis que presumiblemente, a nivel de la proteína, determina un codón de parada prematuro (p.Tyr255*), descrita previamente en la bibliografía como mutación asociada al síndrome de Alagille7,8.
.")

El padre (27 años) presenta facies peculiar (fig. 2), y antecedente de estenosis de rama pulmonar izquierda que precisó intervención quirúrgica en edad pediátrica sin repercusión hemodinámica en la actualidad, y colestasis anictérica (AST/ALT 130/165U/l, GGT 968U/l, FA 442U/l, Bil 0,83mg/dl, Col 210mg/dl) detectada a los 21 años como hallazgo casual en analítica rutinaria atribuida a enolismo. A raíz del diagnóstico del hijo, se realiza colangio-RMN, objetivando una vía biliar extrahepática de escaso calibre con ausencia de visualización de la vía biliar intrahepática y una biopsia hepática que muestra ausencia de ductos biliares en más del 50% de los espacios porta, hipoplasia vía biliar y hemosiderosis intrahepatocitaria. A raíz del diagnóstico de SA en el hijo, se ha diagnosticado también al padre, quién presenta una expresividad menos severa. Cabe destacar la variabilidad de la expresión clínica intrafamiliar, con una afectación hepática importante en el hijo y, en cambio, muy leve en el padre a la edad de 27 años (figs. 1 y 2).
La asociación con atresia intestinal constituye una manifestación que no aparece en la mayoría de las descripciones de este síndrome. Tras una exhaustiva revisión de la bibliografía hemos hallado muy pocos casos que asocien SA a malformaciones intestinales9. El gen JAG1 es uno de los componentes de la vía de señalización NOTCH, la cual desempeña una función primordial en la angiogénesis. La atresia intestinal asociada a SA podría ser consecuencia de una alteración en la vascularización durante el desarrollo embriológico del aparato digestivo10.

