








Clonal natural killer (NK) and T cell expansions induced by Epstein- Barr virus (EBV) and genetic alterations compromising NK cell killing are the most common causes of hemophagocytic lymphohistocytosis (HLH). Generally, HLH is induced by an immune dysfunction where hypercytokinemia develops into reactive hemophagocytosis. In this work we review the causes of HLH and describe a case of a monoclonal expansion of EBV-negative NK cells associated to HLH in a seventeen-month-old girl suffering of Griscelli syndrome type-2 with novel RAB27A mutation and showing partial albinism, persistent fever, hepatosplenomegaly, adenopaties and cytopenias. At diagnosis, no evidence of active viral infections, including EBV, was found. Expansion of NK cells (5300/μl in peripheral blood) CD2+CD7+CD8+CD16+CD56+CD94+CD158a/h+CD158b/j–Perforin+ Granzyme B+ was found. After treatment (HLH-2004 protocol: Cyclosporin, Etoposide and Dexamethasone), NK cell count fell to 850/μl and progressively increased to pre-therapy levels by week 28. X-chromosome inactivation assay demonstrated NK cell monoclonality. NK cells sustained a strong killing and secreted high amounts of IFN-γ. At diagnosis, serum levels of sIL-2R (36,8ng/ml) and IFN-γ (400pg/ml) were elevated. In conclusion, we describe a monoclonal expansion of EBV-negative NK cells highly secretory of IFN-γ as the most probable cause of HLH episode in a patient with Griscelli syndrome type-2. NK cell count recovered normal levels and phenotype after bone marrow transplantation from her HLA identical sister.
Las causas más comunes de linfohistiocitosis hemofagocítica (HLH) son expansiones clonales de células NK y T, inducidas por EBV, así como las alteraciones genéticas que comprometen la actividad asesina de las NKs. Generalmente, HLH se desencadena por una disfunción inmune en la que se desarrolla hipercitoquinemia. En este trabajo se resumen las causas más comunes de HLH y se presenta un caso en el que una expansión monoclonal de células NK, EBV-negativas, se asocia a HLH en una paciente aquejada de Síndrome de Griscelli tipo-2 (GS2). Se trata de una niña de 17 meses con una mutación de nueva descripción en RAB27A, con albinismo parcial, fiebre persistente, hepatoesplenomegalia, adenopatías y citopenias al diagnóstico. No se detectaron evidencias de infecciones virales activas, incluida EBV. Se detectó una expansión de células NKs (5300/μl) CD2+CD7+CD8+CD16+CD56+ CD94+CD158a/h+CD158b/j–Perforin+Granzyme-B+. Tras el tratamiento (Protocolo HLH-2004: Cyclosporina, Etoposido y Dexametasona), la cifra de células NK se redujo a 850/μl y que aumentaron progresivamente hasta alcanzar niveles similares al diagnóstico. El ensayo de inactivación del cromosoma X demostró monoclonalidad de células NK. Dichas células mantenían intacta su actividad asesina y secretaban grandes cantidades de IFN-γ. Al diagnóstico los niveles séricos de sIL-2R (36.8ng/ml) e IFN-γ (400pg/ml) estaban elevados. En conclusión, se describe un caso de una expansión monoclonal de células NK, EBV-negativas, que secretan grandes cantidades de IFN-γ como la causa más probable del episodio de HLH en una paciente con GS2. Tras el trasplante de médula ósea de su hermana HLA-idéntica, las cifras y el fenotipo de las células NK recobraron la normalidad.
Hemophagocytic lymphohistiocytosis (HLH), a prototype of hemophagocytic syndrome, occurs most often in childhood, and commonly involves a persistent unexplained fever, cytopenia, hepatic dysfunction, hepatosplenomegaly, hypofibrinogenemia, and/or hypertriglyceridemia, accompanied by hemophagocytosis in the bone marrow, spleen and lymph nodes. HLH is usually caused by an immune dysfunction whereby hypercytokinemia, produced by activated or clonally proliferating T cells or natural killer (NK) cells and activated macrophages, develops into reactive hemophagocytosis. The serum from HLH patients, especially those infected with Epstein-Barr virus (EBV), contains a high concentration of Th1 cytokines, such as IFN-γ, and TNF-α as well as soluble IL-2 receptor (sIL-2R), but other cytokines such as IL-6 can be also elevated(1).
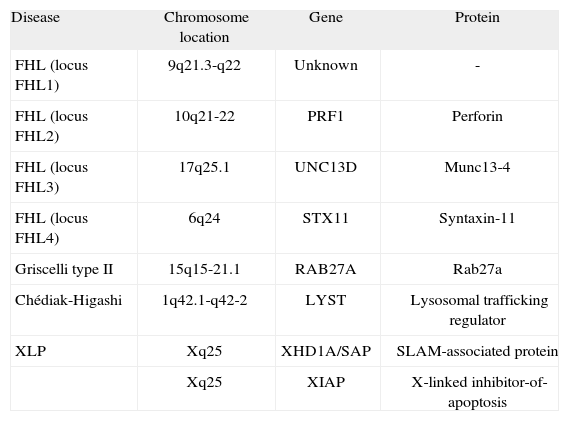
The etiopathogenia of this life-threatening disease is multifactorial. There are familial and non-familial forms of HLH, also classified as primary and secondary HLH, respectively. In the familial form of HLH (FHL), symptoms are usually evident within the first months of life, and less frequently throughout childhood or even into young adulthood. Linkage analysis showed that FHL is linked to four loci: 9q21.3-q22 (FHL1), 10q21 (FHL2), 17q25 (FHL3) and 6q24 (FHL4)(2). While the gene responsible for FHL1 remains unknown, perforin (PRF1) mutations are responsible for FHL2; UNC13D mutations for FHL3 and STX11 mutations for FHL4(2–5) (Table I) Twenty to fifty per cent of FHL cases worldwide are caused by mutations in PRF1. Perforin is a critical protein for cytotoxicity mediated by granules present in NK cells and cytotoxic T lymphocytes, and it is required to fight viral infections as well as to keep the immune system, especially histiocytes, under control. At least three genetic diseases other than FHL have been associated with the hemophagocytic syndrome, including mutations in SH2D1A (X-linked lymphoproliferative disease)(6,7), mutations in CHS1 (Chediak-Higashi disease)(8), and mutations in RAB27A gene (Griscelli syndrome type-II)(9).
Primary haemophagocytic diseases and associated disease-causing genes
| Disease | Chromosome location | Gene | Protein |
| FHL (locus FHL1) | 9q21.3-q22 | Unknown | - |
| FHL (locus FHL2) | 10q21-22 | PRF1 | Perforin |
| FHL (locus FHL3) | 17q25.1 | UNC13D | Munc13-4 |
| FHL (locus FHL4) | 6q24 | STX11 | Syntaxin-11 |
| Griscelli type II | 15q15-21.1 | RAB27A | Rab27a |
| Chédiak-Higashi | 1q42.1-q42-2 | LYST | Lysosomal trafficking regulator |
| XLP | Xq25 | XHD1A/SAP | SLAM-associated protein |
| Xq25 | XIAP | X-linked inhibitor-of-apoptosis |
Extracted from Ménasché G. et al. Immunol Rev 2005;203:165–179.
Griscelli syndrome is a rare autosomal recessive disorder that results in pigmentary dilution of the skin and hair, the presence of large clumps of pigment in hair shafts, and an accumulation of melanosomes in melanocytes. While most patients also develop hemophagocytic syndrome, leading to death in the absence of bone marrow transplantation(10), some show severe neurological impairment early in life without apparent immune abnormalities. Griscelli syndrome type 1 (GS1) is caused by mutation in the MYO5A gene, and presents hypomelanosis with a primary neurological deficit and without immunologic impairment or manifestations of hemophagocytic syndrome(11). Griscelli syndrome with immune impairment, or Griscelli syndrome type 2 (GS2), is caused by mutation in the RAB27A gene. Griscelli syndrome type 3 (GS3), characterized by hypomelanosis with no immunologic or neurologic manifestations, can be caused by mutation in the melanophilin (MLPH). Rab27a is a member of the Rab family of small GTPase proteins. Mutations in the RAB27A gene located on chromosome 15q15-21.1, cause pigment as well as cytotoxic granule transport defects, accounting for the partial albinism and severe immune disorder characteristics of this syndrome. Impaired cytotoxic T lymphocyte (CTL) activity has been clearly established, but NK activity has been scarcely investigated in GS patients. NK function was extensively studied in a patient, possessing a hemophagocytic syndrome with a homozygous Q118X nonsense RAB27A mutation. As expected, CTLs in the patient were not functional and NK cytotoxicity against K562 or 721.221 cells was diminished. Surprisingly, however, CD16- mediated killing was intact in this patient and was therefore RAB27A independent, whereas NKp30-mediated killing was impaired and was therefore RAB27A dependent(12). As previously mentioned, the autosomal recessive immunodeficiencies GS2 and the familial hemophagocytic lymphohistiocytosis type 3 (FHL3) are associated with loss- of-function mutations in RAB27A (encoding Rab27a) and UNC13D (encoding Munc13-4), respectively. However, Rab27a- or Munc13-4-recruitment to lytic granules is preferentially regulated by different receptor signals, demonstrating that individual target cell ligands regulate discrete molecular events for lytic granule maturation. Thus, Munc13-4 deficiency abrogates NK cell release of perforincontaining lytic granules induced by signals for natural and antibody-dependent cellular cytotoxicity (ADCC) through the CD16 molecule. Individual engagement of LFA-1, NKG2D or 2B4 receptors induced degranulation with the intervention of Rab27a, but not Munc13-4(13).
Secondary HLH is associated with a wide variety of diseases such as viral, bacterial and fungical infections, lymphomas and other malignancies, as well as autoimmune and metabolic diseases(14). The second major risk group of childhood HLH is due to EBV infections. More than 50% of pediatric HLH in eastern countries may be associated with EBV infection. Interestingly, most EBV-HLH cases are characterized by mono- or oligo-clonal proliferation of EBV- infected NK or T cells(1). HLH is also seen in patients with peripheral T or NK cell leukemias/lymphomas(15). Again, EBV infection plays a critical role in the pathogenesis of these leukemias/lymphomas, in particular, nasal-type NK/T-cell lymphoma and aggressive NK cell leukemia. NK cell leukemias usually possess a single episomal form of EBV, indicating that this type of leukemia is of clonal origin. Like nasal-type NK/T-cell lymphoma, most patients with NK cell leukemia exhibit an aggressive clinical course. NK cell leukemia is characterized by a systemic proliferation of NK cells, predominantly involving peripheral blood and bone marrow. Besides, it has been reported that EBV-NK leukemia cells constitutively secrete IFN-γ(15), suggesting that IFN-γ may constitute an autocrine survival signal important for the progression of NK cell leukemia, that eventually, might lead to the occurrence of secondary HLH.
A reduced number of HLH cases are associated to systemic inflammatory diseases, which are known as Macrophage Activation Syndrome (MAS)(16,17). Within the systemic inflammatory diseases, HLH most often occurs in Systemic Juvenile Idiopathic Arthritis (JIA), but it can also appear associated to Systemic Lupus Erythematosus (SLE). In contrast to other types of HLH, immune-suppressive therapy, with no bone marrow transplantation (BMT) is generally sufficient to treat these types of HLH(17).
The interest of the case shown in this manuscript, apart from describing for the first time a monoclonal expansion of EBV-negative NK cells associated to HLH in a patient affected for GS2, is that the cosanguinity of parents made necessary to discard or confirm the familial form of HLH, both in the patient and in her HLA identical sister, before BMT could be approved. In addition, it was necessary to demonstrate that the monoclonal expansion of NK cells, highly secretory of INF-γ, could be the origin of the HLH episode in this patient. This manuscript describes the long and intricate analytical procedure necessary for the correct diagnosis, a period during which the life of the patient was seriously at risk. Publication of this case will hopefully contribute to accelerating the diagnostic procedure in future similar cases and to reduce suffering of these patients.
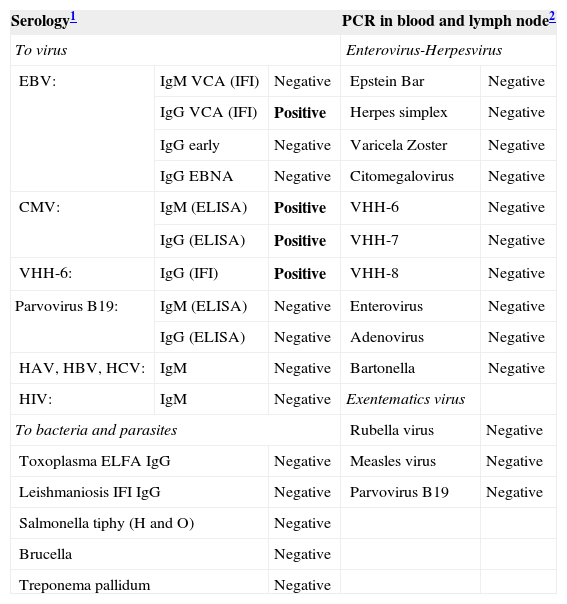
CASE REPORTA 17-month-old girl from Ecuador showing partial albinism was admitted to our hospital in September 2005, because of fever and cytopenias (leukocytes 11.6x109/L, hemoglobin 7.9 g/dL and platelets 65x109/L) persisting over 3 weeks, splenomegaly (10 cm), hepatomegaly (5 cm), multiple adenopathies, purpura and edema (Fig. 1A). Consanguinity (father and mother were cousins and healthy), normal gestation and birth, but recurrent fever episodes from the third month of age were reported. She was vaccinated normally with no remarkable incidences. Biochemical analysis showed normal values for fibrinogen (232 mg/dl, range 150-450), and elevated levels of ferritin (278 ng/ml, normal range 15-150) and triglycerides (311 mg/dl, normal range 50-200). Staphylococcus, coagulase negative, was isolated in one blood culture and treatment with Cefotaxime was set on day 3 after admission; no bacteria or fungi was isolated in urine, blood, bone marrow, lymph node, or spinal fluid afterwards. Serology demonstrated IgG and IgM anti-CMV and IgG anti-HHV-6, as well as a weak positivity of IgG anti-EBV-VCA and negativity for IgM anti-EBV-VCA, and for IgG anti-early and anti-EBNA antigens. No genomic evidences of EBV, CMV, HHV-6, HHV-7, HHV-8, varicela zoster, herpes simple, rubella, measles, parvovirus B19, enterovirus or adenovirus were found either in urine, peripheral blood or lymph node biopsy by PCR analysis (Table II). After admission, she suffered from a rapid clinical and biological deterioration, with persistent fever, food rejection, and worsening of cytopenias (hemoglobin 7.1 g/dl and platelets 7x109/L), visceromegaly and adenopathy. Two weeks after admission, she required transfusion of platelet and erythrocytes. Cytological studies showed an expansion of NK cells both in peripheral blood (5300/μl) and bone marrow aspirate (7540/μl), with a normal expression of perforin and granzyme-B (Fig. 2A) and maintained cytotoxic capacity. Evidence of hemophagocytosis, but not other abnormalities, was observed in the aspirate of the bone marrow (Fig. 1C). Paracortical diffuse hyperplasia in a lymph node biopsy with no signs of hemophagocytosis was informed (Fig. 1D). Genetic studies comprising PRF1, MUNC13-4 and RAB27A demonstrated a novel homozygous mutation, GA transition (c.281G>A), in the coding region of the RAB27A gene (p. Gly97Asp) reported by Dr. Udo Zur Stadt (Hamburg, Germany)(2). In addition, the X-chromosome inactivation assay demonstrated a clear skewed X-inactivation pattern in NK cells compared to patient T cells (Fig. 3). High serum concentration of sIL-2R (36.8 ng/ml) and IFN-γ (400 pg/ml) was additionally detected at diagnosis (Figs. 4A, 4B). Patient was diagnosed as HLH, and treatment set following HLH-2004 protocol (Cyclosporin, Etoposide and Dexamethasone) 2 weeks after admission.
 17-month-old Ecuatorian girl showing partial albinism and suffering from fever and cytopenias persisting over 3 weeks, splenomegaly (10 cm), hepatomegaly (5 cm), múltiple adenopathies, purpura and edema; (B) cell expansion of large granular lymphocytes (LGL cells) in peripheral blood; (C) bone marrow with erithroid, megakaryocytic, and mononuclear-phagocyte hyperplasia and scarce signs of hemophagocytosis. Arrows indicate hemophagocytosis. (D) Paracortical diffuse hyperplasia in a lymph node biopsy with no signs of hemophagocytosis or lymphoproliferative disease; d1 and d2 inserts, interfollicular/paracortical and follicular details of anti-CD3 and anti-CD20 immunohistochemistry analysis, respectively.")
Patient with hemophagocytic lymphohistiocytosis. (A) 17-month-old Ecuatorian girl showing partial albinism and suffering from fever and cytopenias persisting over 3 weeks, splenomegaly (10 cm), hepatomegaly (5 cm), múltiple adenopathies, purpura and edema; (B) cell expansion of large granular lymphocytes (LGL cells) in peripheral blood; (C) bone marrow with erithroid, megakaryocytic, and mononuclear-phagocyte hyperplasia and scarce signs of hemophagocytosis. Arrows indicate hemophagocytosis. (D) Paracortical diffuse hyperplasia in a lymph node biopsy with no signs of hemophagocytosis or lymphoproliferative disease; d1 and d2 inserts, interfollicular/paracortical and follicular details of anti-CD3 and anti-CD20 immunohistochemistry analysis, respectively.
Microbiologic screening at diagnosis
| Serology1 | PCR in blood and lymph node2 | |||
| To virus | Enterovirus-Herpesvirus | |||
| EBV: | IgM VCA (IFI) | Negative | Epstein Bar | Negative |
| IgG VCA (IFI) | Positive | Herpes simplex | Negative | |
| IgG early | Negative | Varicela Zoster | Negative | |
| IgG EBNA | Negative | Citomegalovirus | Negative | |
| CMV: | IgM (ELISA) | Positive | VHH-6 | Negative |
| IgG (ELISA) | Positive | VHH-7 | Negative | |
| VHH-6: | IgG (IFI) | Positive | VHH-8 | Negative |
| Parvovirus B19: | IgM (ELISA) | Negative | Enterovirus | Negative |
| IgG (ELISA) | Negative | Adenovirus | Negative | |
| HAV, HBV, HCV: | IgM | Negative | Bartonella | Negative |
| HIV: | IgM | Negative | Exentematics virus | |
| To bacteria and parasites | Rubella virus | Negative | ||
| Toxoplasma ELFA IgG | Negative | Measles virus | Negative | |
| Leishmaniosis IFI IgG | Negative | Parvovirus B19 | Negative | |
| Salmonella tiphy (H and O) | Negative | |||
| Brucella | Negative | |||
| Treponema pallidum | Negative | |||
 At diagnosis NK cells (black dots) represented 71%, 77%, and 45% of total leukocytes from peripheral blood, bone marrow and the spinal fluid. NK cells showed the following phenotype CD2+ CD3sup- CD3cyt- CD7+ CD8+ CD16+ CD56+ CD94+ CD158a/h+ CD158b/j-/+ Perforin+ and Granzyme B+. Two NK cell subsets could be observed: CD158a/h+CD158b/j+ (5% of total leukocytes) and CD158a/h+CD158b/j- (70% of total leukocytes); (B) discrete lymphocytosis and moderate neutropenia was maintained (left). CD158b/j- NK cell subset dropped from 5300/μl to 850/μl by day 36 due to HLH-therapy (arrow indicates beginning of HLH treatment), and recovered to reach same levels than at diagnosis afterwards. CD158b/j+ NK cell count suffered basically no change. T lymphocytes showed an inverse evolution than NK cells (right).")
NK cell phenotype, and cell counts evolution in time. (A) At diagnosis NK cells (black dots) represented 71%, 77%, and 45% of total leukocytes from peripheral blood, bone marrow and the spinal fluid. NK cells showed the following phenotype CD2+ CD3sup- CD3cyt- CD7+ CD8+ CD16+ CD56+ CD94+ CD158a/h+ CD158b/j-/+ Perforin+ and Granzyme B+. Two NK cell subsets could be observed: CD158a/h+CD158b/j+ (5% of total leukocytes) and CD158a/h+CD158b/j- (70% of total leukocytes); (B) discrete lymphocytosis and moderate neutropenia was maintained (left). CD158b/j- NK cell subset dropped from 5300/μl to 850/μl by day 36 due to HLH-therapy (arrow indicates beginning of HLH treatment), and recovered to reach same levels than at diagnosis afterwards. CD158b/j+ NK cell count suffered basically no change. T lymphocytes showed an inverse evolution than NK cells (right).
![X-chromosome inactivation pattern in T and NK cells. T lymphocytes (CD3+ [A]) and both NK cell subsets (CD3-CD158a/h+, either CD158b/j- [B] or CD158b/j+ [C]) from patient, as well as T lymphocytes (CD3+ [D]) and NK cells (CD3-CD16/56+ [E]) from the mother, were highly purified in a MoFlow sorter (Beckman Coulter). DNA was extracted from purified cells and amplified as described in the methods section. PCR products were digested (+) or not (− ) with Hpall and run in a polyacrylamide gel. A clearly skewed X-chromosome inactivation pattern was observed in both NK cell subsets from patient (B+, C+) compared to patient T cells (A+) and to mother T and NK cells (D+, E+) that used both X-chromosomes.](https://static.elsevier.es/multimedia/02139626/0000002800000003/v1_201305141305/S021396260970037X/v1_201305141305/en/main.assets/gr3.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNdDfbi4GraT4jwPl5dTud2oVGLZONwKWbmHMcYV7eovmDk850OPIJTOWsgDaX/N1BSlnLLDliiOUZr2QN+PiURmEsCLRvGj/G1dU+ApgYkkG6V0JXEp/Ei20Jz4k/0weYrLQObcCK5tjvdRvqU01CAk1klv9N7opIZY0TiPW4Je6UOBHbXUCDoTcWcUaQWBcBfXdtqAmDfd9gENhxiBVzEuzhlluKZ22yM66Hs+b+wAf1HvL0hy9UIwI02l691/YIc= "X-chromosome inactivation pattern in T and NK cells. T lymphocytes (CD3+ [A]) and both NK cell subsets (CD3-CD158a/h+, either CD158b/j- [B] or CD158b/j+ [C]) from patient, as well as T lymphocytes (CD3+ [D]) and NK cells (CD3-CD16/56+ [E]) from the mother, were highly purified in a MoFlow sorter (Beckman Coulter). DNA was extracted from purified cells and amplified as described in the methods section. PCR products were digested (+) or not (− ) with Hpall and run in a polyacrylamide gel. A clearly skewed X-chromosome inactivation pattern was observed in both NK cell subsets from patient (B+, C+) compared to patient T cells (A+) and to mother T and NK cells (D+, E+) that used both X-chromosomes.")
X-chromosome inactivation pattern in T and NK cells. T lymphocytes (CD3+ [A]) and both NK cell subsets (CD3-CD158a/h+, either CD158b/j- [B] or CD158b/j+ [C]) from patient, as well as T lymphocytes (CD3+ [D]) and NK cells (CD3-CD16/56+ [E]) from the mother, were highly purified in a MoFlow sorter (Beckman Coulter). DNA was extracted from purified cells and amplified as described in the methods section. PCR products were digested (+) or not (− ) with Hpall and run in a polyacrylamide gel. A clearly skewed X-chromosome inactivation pattern was observed in both NK cell subsets from patient (B+, C+) compared to patient T cells (A+) and to mother T and NK cells (D+, E+) that used both X-chromosomes.
 and IFN-γ (B) compared to controls (healthy individuals). (C) On day 51 after diagnosis, NK cells and T lymphocytes were highly purified from patient, HLA-identical sister and mother in a Moflow sorter, and 2 million cells per well, stimulated over night with PMA (50 μg/ml) and lonomicyn (0.5 pg/ml) in a 24 well plate. NK cells from the patient secreted 2 to 3 fold more IFN-γ than NK cells from her mother or a HLA identical sister, while T cells from the patient secreted mainly IL-2 but low amounts of IFN-γ.")
Serum soluble-IL-2R and cytokines; in vitro cytokine secretion by NK and T cells. At diagnosis patient showed very high serum levels of sIL-2R (A) and IFN-γ (B) compared to controls (healthy individuals). (C) On day 51 after diagnosis, NK cells and T lymphocytes were highly purified from patient, HLA-identical sister and mother in a Moflow sorter, and 2 million cells per well, stimulated over night with PMA (50 μg/ml) and lonomicyn (0.5 pg/ml) in a 24 well plate. NK cells from the patient secreted 2 to 3 fold more IFN-γ than NK cells from her mother or a HLA identical sister, while T cells from the patient secreted mainly IL-2 but low amounts of IFN-γ.
The patient had an HLA-identical sister that persistently showed reduced NK cell count in peripheral blood (3.25% of lymphocytes) and a barely detectable NK cell cytotoxic activity (< 3% of K562 lysis to 40:1 effector:target cell ratio) in total PBMCs (Figs. 5A, 5B). Due to parent consanguinity, NK cell malfunction was suspected in both patient and sister. However, highly purified NK cells from sister showed normal NK cell phenotype and cytotoxic activity compared to mother's NK cells (Fig. 5C). Though, concluding that the low cytotoxic activity observed in total PBMC from sister was due to a reduced count of rather normal functioning NK cells and, as a consequence, sister was eligible as bone marrow donor. Other family members did not show NK cell alterations either in phenotype or function. Bone marrow transplantation from her sister was carried out successfully in mid-September, 2006 (leukocytes 6.67x109/L, hemoglobin 14.0 g/dl and platelets 370x109/L in June 2008) in Vall d'Hebron Hospital, Barcelona, Spain. Informed consent was obtained from parents and from all members of the family included in the study. Institutional review board approved the analysis of results.
 Cytoplasmic perforin expression in PBL from family members. Numbers indicate the percentage of total NK cells in PBL; (B) cytotoxicity of total PBMC from all family members; and (C) cytotoxicity of purified NK and T cells from the mother and the HLA-identical sister. Lysis of CFSE labeled K562 was measured by flow cytometry using 7-Amino-actinomycin D (7-AAD) staining after 4 hours incubation at 37ºC in a CO2 incubator in the presence of different effector:target cell ratios. All assays included in panels A to C were performed with fresh peripheral blood 5 months after diagnosis.")
Perforin expression and NK cell cytolytic activity. (A) Cytoplasmic perforin expression in PBL from family members. Numbers indicate the percentage of total NK cells in PBL; (B) cytotoxicity of total PBMC from all family members; and (C) cytotoxicity of purified NK and T cells from the mother and the HLA-identical sister. Lysis of CFSE labeled K562 was measured by flow cytometry using 7-Amino-actinomycin D (7-AAD) staining after 4 hours incubation at 37ºC in a CO2 incubator in the presence of different effector:target cell ratios. All assays included in panels A to C were performed with fresh peripheral blood 5 months after diagnosis.
It is important to note that the information about the novel mutation on RAB27A gene of the patient was available more than two years after the bone marrow transplant. So, the diagnosis, treatment and transplantation were based on the clinical and laboratory evidence affordable at that time.
MATERIAL AND METHODSSerological and molecular studies for microbial infectionsPatient sera were tested for the presence of IgM and IgG antibodies against virus, bacteria, and parasites as described in Table II, by using a competitive chemiluminescence immunoassay (CLIA, DiaSorin Laboratories, Anthony, France) at Microbiology Service, University Hospital Virgen Arrixaca (Murcia, Spain). All microbiological molecular studies (see Table II) were performed in the National Center for Microbiology, Instituto de Salud Carlos-III (Madrid, Spain), using properly validated methods.
Routine immunologic studies for HLH screeningIn this section we describe usual immunological studies applied in our laboratory for HLH diagnosis.
- 1.
Soluble IL-2 receptor (sIL2R or sCD25): sIL2R serum concentration is increased during HLH episodes(1).
- 2.
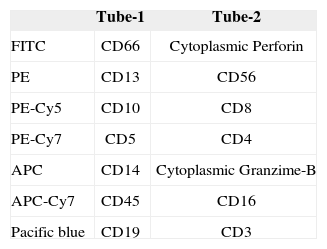
Counting of leukocyte subsets: General analysis of lymphoid and myeloid cell subsets, with particular interest in NK cells. NK cell counts can be reduced in HLH patients(1). In Table III we describe the multicolour flow cytometry antibody panel used in our laboratory.
- 3.
Intracellular expression of Perforin and Granzime-B: Expression of these proteins can be altered in these patients(4).
- 4.
Functional analysis of NK cells: NK cell cytotoxic activity against K562 cell line can be altered in patients with HLH, even in case of normal counts of NK cell and normal expression of Perforin and Granzime-B(4).
PBMC were isolated from heparinized peripheral blood samples by Ficoll-Hypaque (Nycomed Pharma, Oslo, Norway), washed twice, and resuspended in complete medium: RPMI 1640 (Life Technologies, Paisley, Scotland) supplemented with 2 mM L-glutamine (Life Technologies), 50 μg/ml streptomycin, 50 IU/ml penicillin (Flow Laboratories, Irvine, UK), and 10% human AB-serum (Lonza, Verviers, Belgium).
Flow cytometry and cell sortingCytometric analysis was performed on peripheral blood and bone marrow samples with EDTA added as an anticoagulant, as well as in the cerebro-spinal fluid. After collection, samples were immediately stained by a standard immunofluorescence method with different monoclonal antibodies (mAb), acquiring 5x103 lymphocytes in a FACScalibur flow cytometer [Becton Dickinson (BD), San Jose, CA], by setting up a light scatter gate using an anti- CD45-PerCP/CD14-APC mAb combination. Fluorescent mAb, CD2-PE (RPA2.10, BD), CD3-APC (SK7, BD), CD4-PerCP (SK3, BD), CD5-APC (DK23, Dako Diagnostic SA, Denmark), CD7-FITC (M-T701, BD), CD8-PerCP (SK1, BD), CD14-APC (M5E2, BD), CD16-FITC (Leulla, BD), CD19-PerCP-Cy5.5 (SJ25C1, BD), CD25-PE (2A3, BD), CD45-PerCP (HLel, BD), CD56-PE (MY31, BD), CD57-FITC (NK-1, BD), CD94 (NKG2a-purified, kindly provided by Dr. López- Botet), CD158a/h-FITC (EB6, Beckman Coulter, Fullerton, CA, which recognize the KIR2DL1 and KIR2DS1 molecules), CD158b1/b2/j-PE (GK183, Beckman Coulter, which recognize the KIR2DL2, KIR2DS2 and KIR2DL3 molecules), Perforin- FITC (δG9, BD), Granzyme B-FITC (GB11, BD), TCRα/β- FITC (T10B9.1A-31, BD), and TCRγ/δ-PE (Bl, BD), were used at saturating concentration. Simultest IgG1-FITC/IgG2a- Phycoerythrin (BD) and IgGl-TRICOLOR (Caltag, Burlingame, CA) were also used to evaluate background fluorescence. Staining of cytoplasmic antigens (CD3, perforin and granzyme- B) was performed using Intrastain (Dako diagnostic SA, Denmark) following manufacturer's indications.
Absolute number of peripheral blood cell subsets was calculated from the total number of lymphocytes obtained by routine leukocyte count (Coulter T-540, Northwell Drive, England), together with their estimated cytometric percentage values and expressed as cells x103/μl.
To purify total NK cells (CD3-CD16/56+), NK cell subsets (CD3-CD158a/h+CD158b/j- and CD3-CD158a/h+CD158b/j+) and T lymphocytes (CD3+), PBMC were colleted by fluorescence activated cell sorting (FACS) using a MoFlo® cell sorter (Beckman Coulter) equipped with an Argon-ion blue laser (excitation 488 nm) and a red diode laser (excitation 635 nm). Forward and side light scatter and specific fluorescences were used to establish sort regions by using SummitTM software (Cytomation) in a 1–2 drop single cell mode. Purified cells were collected and re-suspended in complete medium.
CFSE/7-AAD cytotoxicity assayNK cell cytotoxic activity was evaluated in a 4 hours assay using K562 cell line (kindly provided by Dr. A. Parrado, Murcia, Spain) as target, and either total PBMC or purified NK or T cells as effectors, following a method previously described with slight modifications(18). Briefly, highly viable K562 cells were stained with 0.25 μM 5, 6-carboxyfluorescein diacetate succinimydyl ester (CFSE, Molecular Probes, Leiden, The Netherlands) for 10 minutes at 37ºC, extensively washed to remove any rest of CFSE and resuspended in complete medium. Effector cells were washed in complete medium and viability of both target and effector cells examined with 0.5% trypan-blue. Effector cells were seeded with a constant number of 50,000 CFSE-labeled target cells at different effector/target ratios 2:1, 10:1, and 40:1 for total PBMC assays, and at 0.4:1, 2:1 and 8:1 for sorting-purified effectors. In parallel, target cells were incubated alone to measure basal cell death. Cells were incubated in a V-bottom 96-well microplates in a total volume of 150 μl of complete medium for 4 h in a 5% CO2 atmosphere at 37ºC. Cell mixtures were then washed in PBS BSA-1% and incubated in the same buffer containing 20 μg/ml 7-amino actinomycin D (7-AAD, Sigma, France) for 20 min at 4°C in the dark. Cell samples were then washed and acquired right afterwards on a FACSCalibur flow cytometer. Mean value of triplicates was used to calculate the percentage of K562 lysis using the following formula: experimental - spontaneous apoptotic target cells.
Cytological and histological analysisPeripheral blood and bone marrow smears were stained with May-Grünwald-Giemsa following standard procedures. A biopsy of an inguinal lymph node was taken at diagnosis. Lymph node was fixed in 10% formalin, and five-micron sections stained with hematoxylin and eosin (H&E) following standard procedures for microscopic examination. Immunohistochemical analysis was done with the following mAb: anti-CD3, anti- CD5, anti-CD20, anti-CD30, anti-CD68, S100 and VS38C, all acquired from Dako Diagnostics (Glostrup, Denmark).
Cytokine and cytokine-receptor measurementSerum sIL-2R was measured with a commercial ELISA (R&D Systems, Abingdom, UK). Cytokines IL-2, IL-4, IL-6, IL-10, IFN-γ and TNF-α were measured by CBA method (BD, San Jose, CA) following the manufacturer's indications both in serum and culture supernatant. Highly purified NK and CD3+ T cell subsets were stimulated over night with 50 ng/ml PMA and 0.5 μg/ml Ionomicyn in complete medium, and supernatants used to measure cytokine secretion by CBA.
Molecular analysis: X-chromosome inactivation assay, TCR gene rearrangement and mutation evaluationGenomic DNA was extracted by using QIAamp DNA Blood Midi Kit (QIAGEN, Hilden, Germany), from highly purified NK cells and T lymphocytes from patient and mother. X-chromosome inactivation analysis was performed by a method previously described(19). Briefly, this method consisted on the analysis of methylation at the androgen receptor locus. PCR is performed on genomic DNA digested with the methylation-sensitive restriction enzyme HpaII, and only the androgen receptor gene residing in the inactivated X-chromosome is amplified. Conditions were the same as those described previously(20).
Molecular analysis of TCR gene rearrangement was performed with DNA extracted from total white cells from peripheral blood and bone marrow by PCR-based TCRgamma gene technique (Master Diagnostica, Granada, Spain). Briefly, PCR amplification of the hypervariable VJ region of the TCR-gamma gene was done following the manufacturer's indications; detection limit was 1-10% of clonal cells, depending on the polyclonal TCR repertoire complexity, and the type of TCR gene rearrangement.
Molecular analysis of perforin (PRF1) and Munc13-4 (UNC13D) mutations were performed by Dr. Udo Zur Stadt (Hematology and Oncology Unit, Ependorff University, Hamburg, Germany), following protocols previously described(2).
HLA class-I and class-II genotypingHLA class I (A and B) typing was performed following a standard complement-dependent microcytotoxicity assay, and using well-defined mAb commercial typing trys (One Lambda, Los Angeles, CA). HLA-C as well as HLA-DRB1 and HLA-DQB1 typing was performed using PCR-singlestrand polymorphism (PCR-SSP) commercial kits (One Lambda, Los Angeles, CA).
RESULTS AND DISCUSSIONDue to parent consanguinity, differential diagnosis of primary/familial versus secondary HLH was aimed from the very beginning of the HLH episode. However, the severity of the clinical symptoms prompted us to establish the right therapy before complete genetic analysis was available. In fact, even though normal status for PRF1 and UNC13D genes was rapidly known, the RAB27A mutation was reported more than 2 years after bone marrow transplantation. Thus, Griscelli syndrome was suspected but not definitively confirmed until RAB27A mutation status was informed. Below we describe the intricate diagnostic procedure, with conclusions reached depending on the clinical and analytical information available at that time.
Familial (FHL) versus secondary HLH diagnosisAccording to the criteria of the Histiocyte Society(21), clinical, analytical and histological evidence (Figs. 1 to 3), together with parental consanguinity, strongly suggested FHL. Most cases of FHL involve mutations of genes implicated in the NK and CTL cytotoxic function. Among them, PRF1 is considered to be altered in 20 to 50% of cases worldwide, although other genes may also be involved(2,3). To confirm FHL diagnosis, NK cell count, perforin expression and NK cytolytic activity in PBMC were evaluated in all family members (Fig. 5B). According to her NK cell counts (71% of PBMC), the patient showed the highest cytotoxic activity (77% of K562 lysis at a 40:1 effector: target cell ratio). Other family members showed an idiosyncratic dispersion of NK cell cytolytic activity, in general in accordance with their NK cell counts. Surprisingly, the HLA-identical sister showed a clear reduction of both NK cell counts (3.25% of PBMC) and NK cell cytotoxic activity (< 3% of K562 lysis at a 40:1 effector: target cell ratio). These last data reinforced the hypothesis of FHL. To clarify this point, NK cell function was evaluated in purified NK cells from the sister. Purified NK cells from the mother, who had shown normal NK cell function, were used as a control (Fig. 5C). This analysis clearly showed that the cytolytic capability of purified NK cells was comparable in mother and sister (54% and 47% of K562 lysis at 8:1 effector: target cell ratio, respectively). The low cytotoxic activity observed in total PBMC from the sister was supposedly due to the reduced count of normally functioning NK cells and, as a consequence, the sister was considered a suitable bone marrow donor.
Additional results, such as: 1) normal intracytoplasmatic expression of perforin in NK cells from all the family members (Fig. 5A), 2) normal intracytoplasmatic expression of granzime-B evaluated in the patient (Fig. 2A), and 3) the absence of mutations in PRF1 and UNC13D reported by Dr. Zur Stadt (Hamburg, Germany) (2), led us to conclude that the patient could be affected by a secondary form of HLH, unless other mutations, that do not alter NK cell function, were involved.
HLH secondary to infection versus monoclonal NK cell expansionThe two major causes of secondary HLH are infection and lymphoproliferative diseases. The former are mainly represented by viruses and, less frequently, by bacterial or even fungal infections. Among the herpes viruses, EBV is a frequent inducer of HLH, but other viruses such as CMV, parvovirus B19, HHV-6, adenovirus and varicella-zoster virus, may also act as inducers(1). Serology only demonstrated positivity to CMV (both IgG and IgM), HHV-6 (IgG) and IgG anti-EBV-VCA. However, none of these or other viruses could be detected by PCR in peripheral blood, bone marrow, lymph node biopsy or urine (Table II). Taken together, the data suggested previous exposure to CMV, HHV-6 and EBV but no acute or active infection at diagnosis that could have induced the HLH.
As regards the second cause of non-familial HLH, cytological studies showed no clear evidence of lymphoproliferative disorders in peripheral blood, bone marrow or lymph node (Fig. 1). However, at diagnosis, the patient showed severe hepatosplenomegaly, and a moderate expansion of monoclonal NK cells (skewed Chromosome X-inactivation) could be detected in blood, bone marrow and even in the spinal fluid. The lack of evidence of lymphoproliferative disease in the bone marrow, suggested that the source of the monoclonal NK cells might be placed somewhere else, such as the spleen or liver. No cytological evaluation of these organs was carried out, so that a lymphoproliferative process in these organs could have further contributed to the severe hepatosplenomegaly, typically observed in HLH. Monoclonal NK cells showed a homogeneous phenotype (Fig. 2A, and data not shown); nevertheless, two different NK cell lines could clearly be distinguished. The first, CD158a/h+CD158b/j+, presented constant cell counts in peripheral blood, but the second, CD158a/h+CD158b/j-, which showed the highest cell count at diagnosis, was dramatically affected by HLH therapy, decreasing after treatment and increasing later on (Fig. 2B). Nevertheless, both NK cell lines showed a similar Xchromosome inactivation pattern (Fig. 3), suggesting the possibility that both lines could have emerged from a common ancestor. However, the CD158a/h+CD158b/j- cell line might carry additional alterations inducing its uncontrolled expansion.
Importantly, no possible NK cell transformation induced by EBV could have occurred, as all molecular studies for EBV were consistently informed negative (Table II). After reviewing the literature, we could not find a single report describing EBV-negative NK cell proliferative disorders or leukemias associated with HLH. Therefore, under unclear diagnosis of lymphoproliferative disorder, and taking into account the absence of EVB infection, we were encouraged to search for other possibilities to explain such expansion of the NK cells. At this point, comparative NK cell phenotype was carried out in all family members (Fig. 6), and the phenotype was related to the HLA-A, -B, and -C phenotype that could be influencing the expression of KIR2DL1 and KIR2DS1 (CD158a/h), KIR2DL2, KIR2DS2 and KIR2DL3 (CD158b/j) and other NK cell associated molecules in each individual. Especial interest was paid to HLA-C phenotypes, in particular to group-1 and group-2 of HLA-C alleles, specific ligands for CD158b/j and CD158a/h receptors, respectively (22,23). In this analysis, no clear relationship between HLA phenotype and the expression of CD158a/h- CD158b/j and CD56-CD57 could be established. However, it can be clearly observed that the NK cell expansion of CD158a/h+CD158b/j- cells was mostly a particular characteristic of the patient.
 are underlined, and group 2 alleles with Lys80 are not. All family members showed similar HLA-DRB1*14 and HLA-DQB1*03 homozygous genotype.")
NK cell phenotype and HLA class-I haplotype in all family members. Dot plots show expression of CD158a/h vs CD158b/j and CD56 vs CD57 antigens on NK cells gated on CD2+CD3- lymphocytes. HLA-A, -B and -C haplotypes are indicated for each member. Group 1 alleles of HLA-C with Asn at position 80 of the α-helix (Asn80) are underlined, and group 2 alleles with Lys80 are not. All family members showed similar HLA-DRB1*14 and HLA-DQB1*03 homozygous genotype.
Once primary forms of HLH, or secondary forms due to infection, were discarded, and even in the absence of clear evidence of lymphoproliferative disease, it was possible to demonstrate that the patient showed high IFN-γ serum levels (Fig. 4B) and, more importantly, highly purified monoclonal NK cells, but not T cells, secreted large amounts of IFN-γ compared with the NK or T cells from the mother and HLA-identical sister (Fig. 4C). It is this condition that, after all, could have induced the HLH episode. Consistent with these observations, it has been suggested that IFN-γ may constitute an autocrine survival signal important for the progression of NK cell leukemia, which might, eventually, lead to the occurrence of secondary HLH(5).
Bone marrow transplantation is the treatment of choice for both FHL and HLH induced by NK lymphoproliferative disease. Fortunately, the HLA-identical sister was available and healthy. After transplant, the NK cell population in the patient was restored to levels that could be considered normal and showed a completely different CD158a/h- CD158b/j profile (Fig. 6).
In summary, this report describe for first time a case of HLH, in a patient with GS2 carrying a novel mutation in RAB27A gene, associated to an expansion of monoclonal EBV-negative NK cells that secreted high amounts of IFN- γ. Importantly, monoclonal NK cells from the patient conserved intact killing capacity. This is an evidence that in vivo NK cells preserved pathways, alternative to Rab27a, for lytic granule maturation, such as the one induced by signals mediated by CD16 and Munc13-4 molecules, for natural and ADCC, as recently described(13). This has direct implications in HLH diagnosis, as not necessarily all primary HLH cases, involving mutation in proteins associated to granule maturation, come out with defective NK cytotoxicity.
ACKNOWLEDGEMENTSThe authors gratefully acknowledge the efforts of all the clinicians and researchers involved in this case, particularly Dr. J. L. Fuster at Pediatric Oncology; Drs. C. Márquez and A. Menasalva of the Microbiology Service, University Hospital Virgen de la Arrixaca (Murcia, Spain), and Dr. A. de la Loma at the National Centre for Microbiology, Instituto de Salud Carlos-III (Madrid, Spain) for their support in the interpretation of all microbiologic analysis; Dr. Lopez-Botet, for kindly provide monoclonal antibodies, Hospital del Mar (Barcelona, Spain); Dr. L. Allende at 12 de Octubre Hospital (Madrid, Spain) for monitoring sIL-2R; and, finally, Dr. Udo Zur Stadt of the Hematology and Oncology Unit, Ependorff University (Hamburg, Germany) for analysis of PRF1 and UNC13D mutations. The authors thank the CIBEREHD scientific program, funded by the Instituto de Salud Carlos III.
DISCLOSURESThe authors declare not having any type of conflicts of interest.