


Self-reactive T cells that escape negative selection in the thymus must be inactivated or eliminated in the periphery. In response to a partial or suboptimal stimulation, T cells become anergic and unable to proliferate and express cytokines in response subsequent re-encounters with antigen. Calcium signaling plays a central role in the induction of anergy, causing the activation of a calcium/calcineurin/NFAT-dependent cell-intrinsic program of self-inactivation. This review will focus on our current knowledge on the mechanisms that regulate the expression of an anergyspecific program of gene expression in T cells, and how the proteins encoded by those genes impose a state of functional unresponsiveness by targeting and modulating the activity of crucial events required for the activation of T cells, which include downregulation of TCR signaling and inhibition of cytokine transcription.
Las células T autorreactivas que escapan al proceso de selección negativa en el timo han de ser inactivadas o eliminadas en la periferia. En respuesta a una estimulación parcial o subóptima, las células T se vuelven anérgicas e incapaces de proliferar y producir citocinas en respuesta a encuentros posteriores con el antígeno. Las señales mediadas por calcio tienen un papel importante en la inducción de anergia, por medio de la activación de un programa de auto-inactivación intrínseco a la célula dependiente de calcio/calcineurina/NFAT. Esta revisión se centra en la descripción de nuestros conocimientos actuales acerca de los mecanismos reguladores de la expresión de un programa de expresión génica específico de la anergia de las células T, y cómo las proteínas codificadas por esos genes imponen un estado funcional falta de respuesta a nuevos estimulos. Esto se lleva a cabo mediante la localización y la modulación de la actividad de sucesos cruciales para la activación de las células T, incluyendo fenómenos como la atenuación de las señales del receptor de linfocitos T (TCR) y la inhibición de la transcripción de citocinas.
A successful adaptive immune system requires the development of the ability to eliminate pathogens, while at the same time it must remain tolerant to self-antigens. This is accomplished through a series of control mechanisms that, for T cells, involve central selection of developing thymocytes in the thymus and peripheral mechanisms that regulate the activity of mature T cells(1-3). Self-reactive T cells that escape negative selection in the thymus have the potential to start dangerous reactions against self-tissues. A series of mechanisms contribute to the establishment of peripheral tolerance, including deletion of self-reactive T cells, antigen ignorance, suppression by regulatory T cells (Tregs) and T cell anergy(4-6).
Exposure to self-antigen in the periphery may lead to cell death. Mature T cells may be eliminated from the periphery through apoptosis induced by antigen-dependent stimulation of self-reactive T cells, in a process that resembles what occurs during negative deletion of thymocytes(4,7,8). Signaling through Fas, which promotes an intrinsic death pathway, Bcl-2 and Bim have been shown to modulate T cell tolerance. Mouse models with defective Bim or Fas and transgenic animals that overexpress Bcl-2 show defects in peripheral T cell tolerance. These mice develop lymphoproliferative responses with increased production of autoantibodies, supporting the role of T cell-deletion in the maintenance of immune tolerance(9-12).
Tregs cells have an important role in the maintenance of the peripheral tolerance against self-antigens. Tregmediated suppression is an active process by which a specific population of T cells is able to regulate the activity of selfreactive T cells(3,6). Suppression by Tregs might be mediated by soluble molecules (e.g. cytokines) and/or require cellto-cell contact(13-17). Tregs can be classified in two major types: natural Tregs, which develop in the thymus and are characterized by the expression of CD4, CD25 and the forkhead family transcription factor Foxp3; and a more heterogeneous group of induced Tregs, that include regulatory T cells type 1 (Tr1), producing IL-10, T helper 3 cells (Th3), which secrete TGF-β, and Foxp3+ iTregs, which may develop in the periphery from naive T cells after antigenic stimulation, under specific conditions(18-22). Although activated human CD4+ T cells may upregulate expression of Foxp3 without acquiring suppressive ability(23), different studies have demonstrated that this transcription factor is the main regulator of Treg development and function(18,20,24,25). Consequently, forced expression of Foxp3 in T cells allows them to acquire regulatory ability(20). Different subtypes of Tregs may use different potential suppression mechanisms, which can utilize inhibitory cytokines such as IL-10, IL-35 and TGF-β, or require cell-to-cell contact(13-17). Cytolysis mediated by granzyme A, in humans, and by granzyme B in mice, has also been implicated in Treg-mediated suppression. Tregs can induce cytolysis on target T cells, in a granzyme dependent-manner(26,27). Some studies have also indicated that Treg cells might induce IL-2 deprivation-mediated apoptosis in responder Foxp3-T cells; however Treg cellmediated suppression of Il-2 mRNA transcription is not restored by addition of exogenous IL-2(28,29). The ectoenzymes CD39 and CD73 can induce production of pericellular adenosine, which activates the adenosine A2A receptor and suppresses effector T cell functions. In addition, A2A engagement also promotes Treg generation by inhibiting IL-6 expression(30). T cells can also be suppressed by transfer of the inhibitory second messenger cyclic AMP from Tregs(31). Suppression has also been shown to respond to targeting of dendritic cells. Dendritic cells interact with surface molecules expressed by Tregs, such as CTLA-4, LAG-3, through CD80/CD86 and MHC Class II, respectively, leading to an inhibition of their capacity to activate conventional T cells(14,17,32). Finally, it has been suggested that Tregs can induce the production of indoleamine 2,3 dioxigenase (IDO), an immunoregulatory tryptophan-degrading enzyme ondendritic cells (33,34). A better understanding of Treg-mediated tolerance may come from a more detailed characterization of how different types of Tregs may exert their suppressive activity and how the context in which suppression takes place may modulate Treg function and development.
T cell anergy is also a mechanism of peripheral tolerance in which lymphocytes are functionally inactivated following antigen encounter(35-37). Recognition of antigen (signal 1) in the absence of co-stimulation (signal 2) causes T cells to enter an anergic state, defined by defective activation in response to antigen re-encounter, which translates in decreased proliferation and defective production of IL-2 and other cytokines(38-40). Engagement of the CD28 costimulatory receptor seems to have a pivotal role in preventing the induction of clonal anergy, either through a direct effect, inhibiting the synthesis or function of anergic factors; or through an indirect effect, by inducing the production of IL-2(4i-43). Although this "2 signal" model provides a simple mainframe to understand how clonal anergy is induced, in vivo T cells are likely to receive many different signaling inputs. The balance between costimulatory signals and inhibitory signals determines the nature of the response of T cells to antigen, leading to immunity or anergy. For instance, CD28 and ICOS co-receptors provide positive signals that promote and sustain T-cell responses(44-46); while CTLA-4 and PD-1 are negative co-receptors that limit responses, controlling the extent of the immune response and protecting against the development of autoimmune diseases(47-54). It is therefore the integration of positive and inhibitory signals that controls the establishment of an anergic tolerant status in vivo in T cells.
A different model of in vivo anergy has been described that occurs following transfer of T cell receptor (TCR) transgenic T cells into a T cell-deficient mouse that expresses the cognate antigen for the transferred T cells. These T cells become unresponsive, but as opposed to the classical clonally anergic T cells, they require permanent antigen exposure to remain anergic. Furthermore, signaling blockade occurs upstream of Ras and seems to respond to defective zeta chain-associated protein of 70 KDa (Zap-70) activation. This state has been termed adaptive tolerance(55-57). Although we will focus our review on the current knowledge on the mechanisms that regulate clonal anergy in vitro and in vivo, it is likely that different anergy-inducing mechanisms may be activated depending on the specific context in which the anergizing stimulus is delivered.
SIGNALING CASCADES THAT INDUCE T CELL ANERGYComplete T cell activation requires recognition through the antigen specific TCR of peptides presented by selfhistocompatibility complex (MHC) molecules on antigen presenting cells, in addition to a co-stimulatory signals, as the one delivered by CD28. As discussed above, stimulation of T cells by engagement of their antigen receptor in the absence of co-stimulation leads to clonal anergy(1,37). This partial activation does not efficiently activate mitogenactivated protein kinases (MAPK), or the phosphoinositide 3-kinase/protein kinase B (PI3K/AKT) and IkB-kinase (IKK) signaling pathways, which leads to changes in the genetic program expressed in those T cells. Instead, a specific anergyassociated program of gene expression is induced(43,58,59). This situation can also be caused by chronic exposure to antigens or in response to a high-dose soluble antigen(60). High levels of extracellular adenosine, which may be released by tumor cells or Tregs, can also induce anergy in CD4+ T cells(30,61,62). Whether these conditions also result in limited co-stimulatory signaling remains to be determined.
In T cells, engagement of the TCR complex induces a conformational change that makes the cytosolic tails of the different CD3 chains accessible to phosphorylation(63). TCR clustering promotes the recruitment of the CD4 or CD8 co-receptors and the associated src-family kinase Lck, which phosphorylates immunoreceptor tyrosine based activation motifs (ITAMS) on the CD3 chains(64,65). Docking sites are then created for Zap-70, which binds through its tandem src homology 2 (SH2) domains and is also phosphorylated by Lck(66,67). Zap-70 phosphorylates the membrane-associated linker for T-cell activation (LAT) and the SH2 domain-containing leukocyte protein 76 KDa (SLP-76), which is recruited with Grb2-related adapter downstream of Shc (GADS)(68). These adaptor proteins create a docking site for the phospholipase C-γ1 (PLCγ1), which is activated by the Tec family kinase Itk. PLCγ1 cleaves the membrane phospholipid phosphatidylinositol-4, 5-biphosphate (PIP2) into inositol 1,4,5-triphosphate (IP3) and diacylglycerol (DAG). DAG diffuses laterally in the plasma membrane to activate Ras and the MAPK pathway, likely through activation of RasGRP1. IP3 induces the aperture of calcium channels in the endoplasmic reticulum, which releases intracellular calcium stores. The subsequent increase in the intracellular calcium concentration levels leads to the opening of calcium-released activated calcium channels (CRAC) in the plasma membrane(69,70). While full activation of MAPK and IKK requires costimulation through CD28, intracellular calcium increases required for optimal activation of the calcineurin/ Nuclear factor of activated T cells (NFAT) axis are attained through TCR engagement(71). As we will discuss below, this unbalanced activation of calcium signaling is responsible for the induction of the anergic state in T cells(1,43,72,73). Establishment of hyporesponsiveness in anergic T cells may also result from a lack of activation of the mammalian target of rapamycin (mTOR)(74,75). mTOR is an atypical serine/threonine protein kinase that regulates cell growth and proliferation, gene transcription, mRNA turnover and translation, ribosomal biogenesis, vesicular trafficking, autophagy, cytoskeletal organization, cell size and mammalian development(76). mTOR is activated PI3K and AKT through TCR/CD28/IL-2 receptor co-stimulation, and it has been shown to play a crucial role in the regulation of T cell function(77). mTOR activity has been proposed to be required to avoid the establishment of an anergic state(78). Consequently, T cells activated in the presence of the mTOR inhibitor rapamycin become anergic even when costimulatory receptors are engaged(79). This effect is independent of the regulatory effect of mTOR on the cell cycle, as activation in the presence of cell cycle inhibitors that do not target mTOR does not result in anergy(80). Anergy in T cells can be reversed by signaling through the IL-2 receptor. This effect is independent of CD-28 costimulation, since exogenous IL-2 can reverse anergy in the absence of CD28 engagement(41). lL-2 signaling also prevents the expression of genes that are crucial in the establishment of anergy in T cells, such as DGKα, Caspase-3, Ikaros, Cbl-b and Grail, and this effect is blocked by the presence of the mTOR inhibitor rapamycin(42). All these data suggest, thus, that mTOR acts a sensor of environmental signals in T cells that determines whether they will be activated or become anergic(75,78). In addition, mTOR is also involved in regulating the fate of T cells leading to differentiation into effector cells or Tregs, which in turn are anergic. Under normal activating conditions in the absence of TGF-β, T cells lacking mTOR differentiate into Foxp3+ Tregs, but are not able to differentiate into effector T helper 1(Th1), Th2, or Th17 cells(74). The PI3K/AKT/mTOR signaling pathway has been shown to regulate Foxp3 expression, since its inhibition induces the expression of this transcription factor in activated CD4+ T cells(81). Moreover, the expression of a constitutively active AKT impairs Foxp3 upregulation(82). There is evidence that of the AKT/mTOR signaling pathway can inhibit the activation of Smad3(83), providing a possible mechanism as to how mTOR activity may be able to dowregulate Foxp3 expression.
TRANSCRIPTIONAL REGULATION OF T CELL ANERGYDespite the possibility that different forms of anergy may exist, different studies have characterized that the induction of clonal anergy in CD4+ T cells is dependent on NFAT transcription factors. Analysis of the phenotype of mice lacking different NFAT family members supports the crucial role that NFAT transcription factors have in the regulation of T cell tolerance(84). Mice that lack NFAT1 and/or NFAT4 develop hyperproliferative disorders with defects in the mechanisms that regulate T cell inactivation and anergy(85-87). As described above, TCR engagement in absence of co-stimulation, leads to activation of calcium signaling with limited Ras-MAPK and IKK activation. Increased levels of intracellular calcium promote the activation of the calcium/calmodulin-dependent phosphatase calcineurin. Calcineurin can then dephosphorylate NFAT proteins, which are present in the cytosol in a highly phosphorylated state. Dephosphorylated NFAT proteins translocate into the nucleus, where they can cooperate with other transcription factors to induce the activation of specific programs of gene expression(71,88,89). NFAT activity is mainly regulated by its subcellular localization. NFAT nuclear localization is a net result of the rate of nuclear import/export, which is regulated by means of interplay between calcineurin and NFAT kinases. NFAT is maintained in the cytosol in a hyperphosphorylated state by cytosolic maintenance kinases. In the nucleus, it can be rephosphorylated by nuclear kinases, which regulate the retention and shuttling out of NFAT proteins(89-99). NFAT proteins are also subject to regulation through other mechanisms, which might contribute to modulate the expression of NFAT-dependent genes. Sumoylation of NFAT1 has been identified as a nuclear retention mechanism, and it might also regulate NFAT transcriptional activity(100). Sumoylation of the isoform C of NFAT2 induces relocalization to promyelotic leukemianuclear bodies and promotes recruitment of histone deacetylases (HDCACs), which results in repression of the Il2 promoter(101). Additionally, poly-ADP-ribosylation can also regulate NFAT nuclear export and NFAT-dependent transcriptional activity, suggesting that Poly(ADP-ribose) polymerase-1 could act as a corepressor or coactivator of NFAT, as it has been described for other transcription factors(102-104). NFAT proteins have also been shown to cooperate with transcriptional repressors, such as ICER, a member of the CREB/CREM family of transcription factors, PPRγ and P21 SNFT(102,104). However, it is not known whether these complexes may cooperate with NFAT in the regulation of T cell inactivation and therefore in the induction of anergy.
During T cell activation, induction of NFAT and Activator Protein (AP)-1 transcription factors cooperatively regulates the expression of T cell activation-associated genes(105-108). However, there is ample evidence that shows that in response to tolerizing stimuli CD4+ T cells induce a specific set of genes which are different from those expressed in activated T cells(1,43,58,59). The transcription of these genes is NFAT1-dependent, since in Nfat1−/− T cells and T cells treated with the calcineurin inhibitor cyclosporine fail to induce their expression and do not become anergic in response to anergizing stimuli(43). The expression of NFAT-dependent anergyassociated genes is responsible for the induction of an unresponsive state and for the inhibition of cytokine expression in anergic T cells(72,109–116).
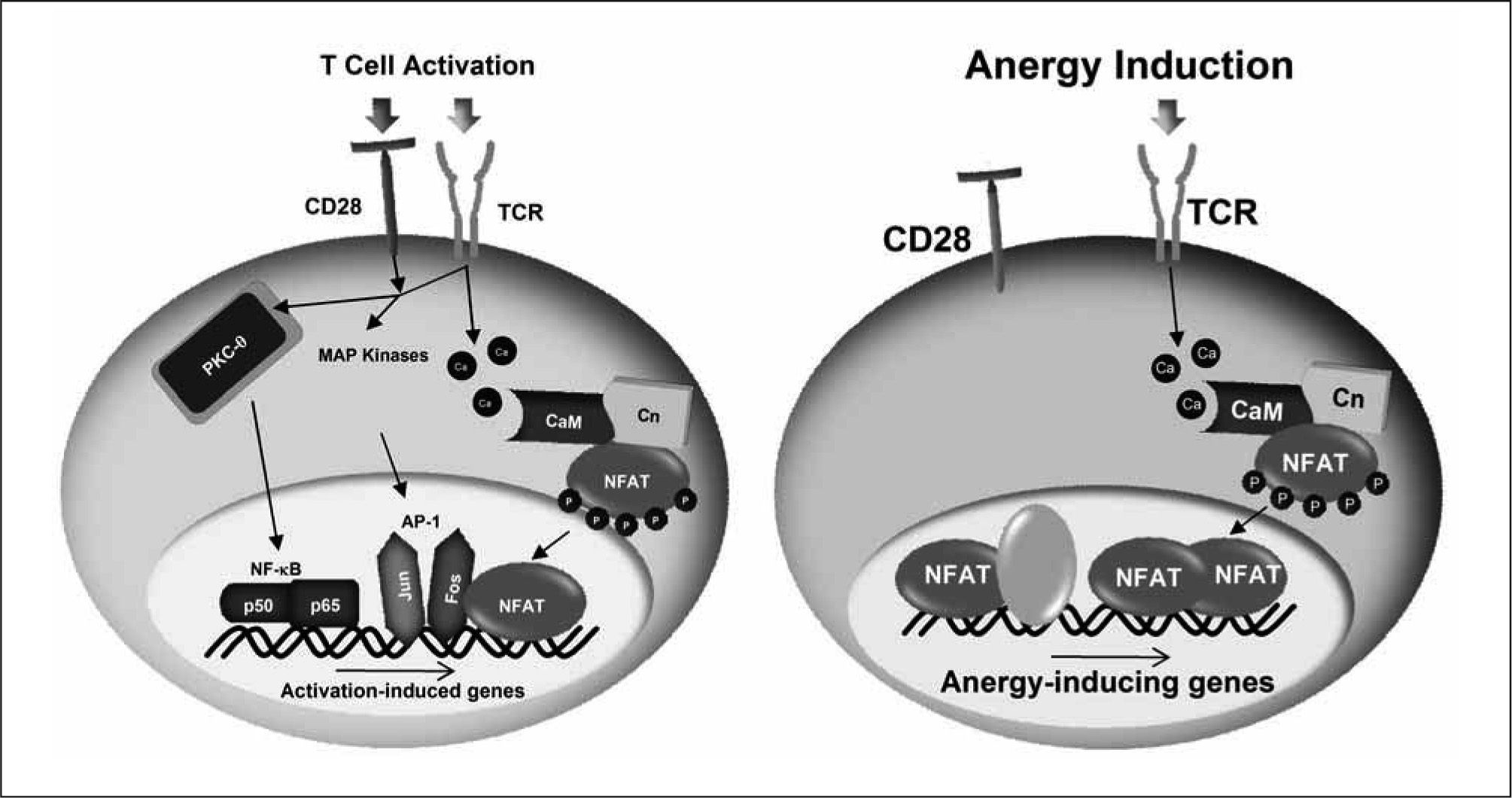
Although NFAT may cooperate with other transcription factors to induce the expression of some of the genes expressed in T cells in response to an anergizing stimulus, we have recently shown that NFAT1 dimers are the transcriptional complexes that regulate the induction of many anergyassociated genes in T cells. NFAT dimers bind to κB-like sites, containing two tandem consensus NFAT-binding sites separated by one or two nucleotides(117-119). The importance of NFAT dimers in the expression of anergy-inducing genes is supported by the fact that mutant NFAT proteins unable to form dimers are incapable of inducing the expression of several anergy-associated genes and fail to restore the susceptibility to anergizing stimuli in Nfat1−/− T cells(119). Furthermore, we have found NFAT dimer sites in the proximal promoter Grail, an E3 ubiquitin ligase which has a crucial role in the regulation of T cell anergy, that when mutated prevent the calcium-induced expression of this gene(119). The nature of the NFAT-containing transcriptional complexes determines, thus, the outcome of the T cell response to antigen: activation if, as a consequence of concomitant engagement of the TCR and costimulatory receptors, NFAT/AP-1 complexes form and bind to the promoters of genes involved in a productive T cell response; or anergy if, in response to a suboptimal stimulation, inefficient AP-1 activation occurs leading to the formation of NFAT homodimers, which induce the expression of anergy-inducing genes (Figure 1). Recruitment of NFAT and AP-1 composite sites is likely to prevent formation of the much lower affinity NFAT homodimers on κB-like sites, thereby preventing the expression of anergy-associated genes(1).
 /calcineurin (Cn). Those signaling pathways lead to the activation of a series of transcription factors, including NF-κB (p50, p65), AP-1 (Fos, Jun) and NFAT. Under these conditions, NFAT forms transcriptional complexes with AP-1 on composite sites located in the promoters of many cytokine genes and induces their expression. In the absence of costimulation, AP-1 proteins are not fully activated and instead NFAT dimers are formed that direct the expression of anergy-associated genes. It is also likely that transcriptional complexes formed by NFAT and other transcription factors may contribute to the expression of some of the anergy-inducing genes.")
Distinct NFAT-containing transcriptional complexes regúlate T cell activation and anergy. Engagement of the TCR and CD28 induces the activation of different signaling cascades that include among others PKC-, MAPKs and calcium/calmodulin (CaM) /calcineurin (Cn). Those signaling pathways lead to the activation of a series of transcription factors, including NF-κB (p50, p65), AP-1 (Fos, Jun) and NFAT. Under these conditions, NFAT forms transcriptional complexes with AP-1 on composite sites located in the promoters of many cytokine genes and induces their expression. In the absence of costimulation, AP-1 proteins are not fully activated and instead NFAT dimers are formed that direct the expression of anergy-associated genes. It is also likely that transcriptional complexes formed by NFAT and other transcription factors may contribute to the expression of some of the anergy-inducing genes.
Other transcription factors may also be implicated in the expression of anergy-inducing genes. Early growth response 2 and 3 (Egr-2/ Egr-3) are upregulated in response anergic stimuli and regulate the expression of inhibitory proteins that promote anergy(115). The expression of Egr2/Egr-3 is in turn regulated by NFAT. These transcription factors can also suppress Egr-1 and NAB2 expression, which cooperate to induce IL-2 production. This may result in the inhibition of Il2 expression as a previous step to the establishment of an anergic state(115,120,121).
ANERGY-ASSOCIATED PROTEINS MAINTAIN THE UNRESPONSIVE STATE IN ANERGIC T CELLS THROUGH THE INHIBITION OF TCR SIGNALINGAnergic T cells upregulate an NFAT/calcineurindependent anergic program of gene expression. Proteins encoded by anergy-associated genes can directly inhibit signaling pathways activated by TCR and/or CD28 engagement, and cause suppression of T cell responses to subsequent antigen encounters and the maintenance of an anergic state(73,122–125). Anergic T cells express at least three different E3 ubiquitin ligases(72,109,111). In a model of clonal anergy, some of these ligases translocate to the plasma membrane and target specific proteins that positively regulate T cell activation for ubiquitination and degradation(72,126–128). Although the mechanisms underlying the role of ubiquitination in the maintenance of T cell anergy are still not completely characterized, recent studies have unveiled that these E3 enzymes play an essential role in the anergy-associated down-modulation of TCR signaling.
Egr2 and Egr3 induce the expression of Cbl-b, a RINGtype E3 protein, in anergic T cells(115). The involvement of this enzyme in T cell anergy is underscored by the fact that the loss of Cbl-b impairs induction of T-cell tolerance both in vitro and in vivo, and mice deficient in this E3 ligase develop spontaneous signs of autoimmunity(111-129). While several targets have been identified for the ubiquitin ligase activity of Cbl-b(72,130–133), after receiving an anergizing stimulus, T cells that do not express this enzyme show reduced inactivation and degradation of PLC-γ1 and slower kinetics of synapse disintegration than control anergic T cells(72,111). Interestingly, Cbl-b has also been shown to regulate anergy in B and NKT cells(133,134)
Grail is also a RING-type E3 protein, which is expressed in anergic T cells. Upregulation of Grail expression is caused by binding of NFAT dimers to two κB-like NFAT dimer sites located in its proximal promoter(119). Grail is crucial for the establishment of T-cell tolerance both in vitro and in vivo(109,112,135,136). Grail causes deficient activation of RhoA by targeting for ubiquitination and stabilizing the Rho guanine dissociation inhibitor (RhoGDI)(127). Grail has also been reported to bind and ubiquitinate several transmembrane proteins, including the tetraspanin superfamily members CD81 and CD151 and the costimulatory molecule CD40L, targeting them for degradation(126,128,137). In addition, overexpression of Grail has been shown to impair actin cytoskeletal organization negatively affecting T cell-antigenpresenting cell interactions(138). The importance of Grail in T cell tolerance has been supported by a recent study that has characterized a Grail-deficient mouse model. T cells from this mouse are hyperresponsive and resistant to anergy induction in vivo(112).
The E3 ligase Itch was first characterized in studies of a spontaneous mutation that produced mice with constant itching of the skin and induced the development of autoimmune disease(139). This HECT-type E3 ligase is also expressed in anergic T cells. Upon restimulation, Itch translocates into detergent-insoluble microdomains in the plasma membrane, where it can ubiquitinate PLC-γ1 and PKC- recruited to the TCR signalosome. Ubiquitination of these proteins sorts them into the endocytic pathway where they are eventually degraded by the lysosomes(72). Itch can also target Jun family transcription factors (e.g. JunB and c-Jun), although the relevance of this process to anergy in T cells is still not known(140). Similar to Cblb, Itch-deficient T cells show impaired induction of anergy(72).
How the function of these E3 ligases may integrate to induce hyporesposiveness in anergic T cells, and the specific targets responsible for that effect remain yet to be determined.
Deltex1 (DTX1) has also been recently shown to regulate T cell activation and tolerance. As an E3 ligase, it may be responsible for the downregulation of the MAP kinase ERK kinase kinase 1 (MEKK1)(141). Expression of DTX1 is regulated by NFAT in anergic T cells, where, independently of its ubiquitin ligase activity, it regulates the expression of Cbl- b and the growth arrest and DNA-damage-inducible 45, (Gadd45,) through cooperation with Egr2(142).
Clonally anergic T cells were initially characterized by their impaired ability to activate Ras signaling(143). Recent evidence indicates that this impairment may be explained, at least in part, by the conversion of DAG into phosphatidic acid mediated by diacylglycerol kinase-α (DGK-α)(113,116). This kinase is expressed in a NFAT-dependent manner in anergic T cells(43,113). Decreased levels of DAG caused by increased expression of DGK-α prevent activation of the Ras-dependent ERK/MAPK signaling cascade. Accordingly, in vivo induction of T cell anergy in mice deficient in DGK-α is greatly impaired(113,116).
Our laboratory has discovered that Caspase-3, another calcium/NFAT-dependent gene which is upregulated in anergic T cells(43), is required for the induction of T cell unresponsiveness(114). Caspase-3 is a crucial effector of apoptosis and other cell functions in the immune system(144-154). In anergic T cells, Caspase-3 is likely activated by calciumactivated calpains, and recruited to the plasma membrane, where it cleaves and inactivates GADS and the guaninenucleotide exchange factor Vav1, blocking TCR signaling. Furthermore, pharmacologic inhibition or genetic deletion of Caspase-3 in T cells makes them resistant to anergizing stimuli. The inability of activated Caspase-3 to induce cell death in anergic T cells is likely due to a tightly controlled subcellular compartmentalization of Caspase-3, which is restricted to the plasma membrane, preventing access to pro-apoptotic substrates(114).
Recently, the type III histone deacetylase Sirt1 has been shown to be essential for maintenance of T cell tolerance in mice. Sirt1 is expressed in anergic T cells where it binds and deacetylates c-Jun, which results in the inactivation of this transcription factor(155). The importance of Sirt1 in the maintenance of T cell tolerance is underscored by the fact that T cells from mice lacking Sirt1 are hyperresponsive and resistant to the induction of anergy in vitro, which translates in vivo in the development of autoimmunity in these animals(155).
Calcium signaling has also been shown to be responsible for an impaired activation of LAT. This defect seems to be a consequence of decreased recruitment of this adaptor protein to the immunological synapse caused by defective palmitoylation(156). It should prove very valuable to identify the mechanisms that regulate the activity of the palmitoyl acyl transferases responsible for this defect in anergic T cells.
ANERGY-INDUCING PROTEINS ALSO MAINTAIN AN UNRESPONSIVE STATE IN ANERGIC T CELLS BY SILENCING CYTOKINE GENE EXPRESSIONOne of the consequences that ensue after T cells receive tolerogenic stimuli is a marked defect in the capacity of those cells to produce and secrete cytokines when new antigen is encountered. This impairment is not only the result of the inhibition of signaling pathways downstream of the TCR, but recent reports have clearly established that cytokine expression in anergic T cells is also regulated at the transcriptional level. Transcriptional repressors are responsible for the active suppression of cytokine gene expression. Furthermore, binding of those repressors induces epigenetic modifications that may be responsible for the long-lasting nature of the anergic phenotype(5,84).
Changes in the expression of the Il2 gene have been shown to correlate with specific epigenetic changes. As T cells develop and differentiate, they acquire the capacity to respond to antigen with increasing ability to produce IL-2. This increased ability to express IL-2 is associated with increased histone acetylation of the Il2 locus. This effect cannot be achieved merely by TCR engagement and requires costimulatory signals, likely through CD28(157). Partial activation of T cells does not only result in a passive loss of histone acetylation on the Il2 promoter, but an active mechanism of chromatin remodeling is engaged. Several HDACs that modify this locus are recruited and prevent histone reacetylation in response to restimulation, even in the presence of costimulation, in anergic T cells(110,158). Ikaros expression is upregulated in a calcium/NFAT signaling-dependent manner during anergy induction(43,110). Ikaros is the founding member of a kruppel-like zinc finger family of transcription factors that have key roles in the regulation of lymphocyte development(159-162). In anergic T cells, Ikaros directly binds to the Il2 promoter and induces histone deacetylation on this locus, a modification associated with silent chromatin (Figure 2)(110,158). Supporting the crucial role of this transcriptional factor in T cell anergy, T cells deficient in Ikaros or expressing a dominant-negative form of this transcription factors become resistant to anergy(110,158).
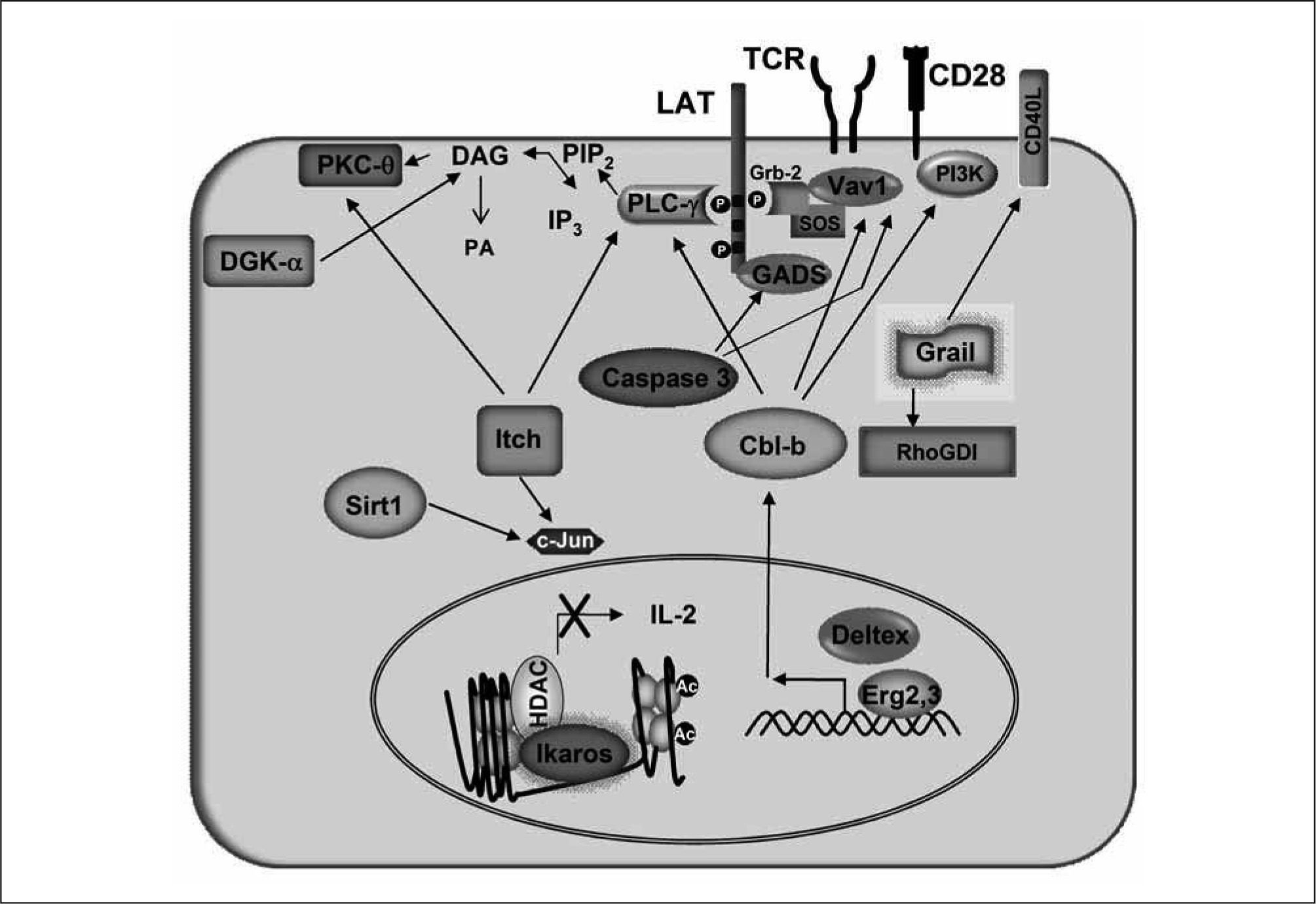
, preventing activation of DAG-dependent signaling downstream of the TCR. In anergic T cells, Ikaros binds to the Il2 promoter where it recruits HDACS that induce epigenetic changes in this locus, which lead to repression of Il2 expression.")
Proteins encoded by anergy-associated genes are responsible for blockingsignaling downstream of the TCR and repressing cytokine expression in anergic T cells. Upregulation of the expression of Cbl-b is induced in anergic cells by Egr2 and Deltex. Cbl-b-mediated ubiquitination ofVavl, PLC-γ1 and PKC- regulates their function and degradation. Cbl-b can also bind the p85 subunit of PI3K and inhibit its recruitment by CD28. Itch also targets PLC-γ1 and PKC- for ubiquitination and subsequent degradation, and it can also downregulate Jun activity. Grail has been reported to stabilize RhoGDI through ubiquitination, which ultimately interferes with TCR-induced actin cytoskeleton reorganization. Grail also regulates the turnover of membrane associated receptors such as CD40L. The HDAC Sirt1 deacetylates and inactivates cJun in anergic cells. Caspase-3 cleaves and prevents the activation-induced membrane-recruitment of GADS and Vav1. DGK transforms DAG into phosphatidic acid (PA), preventing activation of DAG-dependent signaling downstream of the TCR. In anergic T cells, Ikaros binds to the Il2 promoter where it recruits HDACS that induce epigenetic changes in this locus, which lead to repression of Il2 expression.
Expression of IL-2 in activated T cells also correlates with DNA demethylation on the proximal promoter of this cytokine(163). In anergic T cells, increased DNA methylation of the Il2 promoter has also been shown to enforce downregulation of Il2 expression in an in vivo model of superantigen-induced T-cell anergy. These results further confirm that cytokine expression is modulated by epigenetic changes that are actively induced and maintained in anergic T cells(164).
Other mechanisms involving transcriptional repression have been also proposed to account for the inhibition of Il2 expression in anergic T cells. CREM/CREB complexes and p50 homodimers bind the Il2 promoter and may contribute to silencing its expression in a number of anergy models(165,166). Tob, a member of the Tob and BTG antiproliferative protein family, is also upregulated in anergic T cells and contributes to the repression of Il2 expression in these cells(167). As described for Ikaros(158), Tob is also involved in enforcing the costimulation requirement in naive T cells. The function of Tob is likely mediated through enhanced binding of Smad repressors to the Il2 promoter(167). Interestingly, Smad3 also binds and silences the Il2 promoter in anergic cells, responding to increased levels of the cell cycle regulator p27kip(168).
ANERGY IN TREGSTregs are also anergic. Foxp3+ Tregs do not produce IL-2 when activated with anti-CD3 and anti-CD28(58,169,170). As we discussed before, a major factor that regulates the development and function of Tregs is the forkhead wingedhelix transcription factor Foxp3(18,20,171,172). Foxp3 expression in Tregs depends on NFAT, since NFAT2 can cooperate with Smad3 to induce Foxp3 gene expression in response to signals received trough the TGF-β and the IL-2 receptors(173). Recently, complexes of the transcription factors RunX and CBF, have also been shown to be essential in the maintenance of Foxp3 expression in Tregs(174). Foxp3 binds to the regulatory regions of the Il2 and Ifng loci, suppressing the production of IL-2 and IFN-γ in Tregs(175). Silencing of these cytokine genes responds to the ability of Foxp3 to recruit chromatin remodeling complexes containing histone deacetylases(176). Indeed, the chromatin at the Il2 locus in Tregs has been shown to present a closed conformation that is not altered when those cells are activated with anti-CD3 and anti-CD28(177). Foxp3 can form complexes with NFAT1 that bind to the Il2 promoter. This interaction displaces AP-1 and prevents it from forming activating complexes with NFAT at the NFAT/AP-1 composite sites located on the Il2 promoter. This mechanism may also contribute to prevent Il2 expression in Tregs(178). Additionally, Foxp3 can also directly interact with c-Jun, preventing binding of AP-1 complexes to their targets, including the Il2 promoter(179). Interestingly, Foxp3 and NFAT1 interactions not only control IL-2 expression, but also Treg function. Mutations that prevent this interaction lead to loss of suppressive capability of Tregs both in vitro and in a mouse model of autoimmune diabetes(178). These results might indicate that anergy and suppressive activity are coupled in Tregs. Alternatively, they may just indicate that NFAT/Foxp3 complexes regulate the expression of many other target genes in Tregs. It remains yet to be determined if the mechanisms that regulate unresponsiveness in anergic T cells are also engaged in Tregs. In support of this possibility, Grail expression is also upregulated in Tregs, and in vitro overexpression of Grail confers T cells a regulatory phenotype(180). Recently, it has been reported that Eos, a member of the Ikaros family of transcription factors, interacts with Foxp3 and promotes chromatin modifications, which mediate silencing of Foxp3-dependent genes in Tregs(181). Therefore, similar to the role that Ikaros plays in anergic T cells, Eos also seems to have a critical role in regulating gene transcription in Tregs and in modulating their suppressor activity. Recent work has suggested a possible role for NFAT proteins in the regulation of Treg-mediated suppression. Grail, a NFAT-dimer dependent gene expressed in anergic T cells, is also upregulated in CD4+ T cells when suppressed by Tregs(182). Moreover, T cells that lack NFAT1 and NFAT4 become resistant to suppression by Tregs(183). These results suggest, thus, a central role for NFAT in anergy, and in the Treg-mediated dominant form of tolerance.
CONCLUDING REMARKSThe last few years have seen important advances in our understanding of the complex molecular mechanisms that regulate the induction and maintenance of anergy in T cells. Unchecked calcium signaling that results from a partial stimulation leads to the activation of NFAT proteins but minimal AP-1 activation, which requires costimulation. In the absence of other activation-induced transcriptional partners, a NFAT-dependent program of gene expression is engaged. A series of studies have identified and characterized many of the negative regulators of T cell activation induced by calcium/NFAT signaling in anergic T cells. NFAT dimers may play a crucial role in the regulation of the expression of anergy-associated genes. These include, among others, E3 ubiquitin ligases, caspase 3 and DGKα, which inhibit T cell function by blocking TCR-activated proximal signaling events; and Ikaros, which represses Il2 transcription. In vivo, T cells are likely to also receive input from the engagement of other negative regulatory receptors, such as CTLA-4 or the A2A receptor, which may contribute to the establishment of an unresponsive state in self-reactive T cells. How those signals may be integrated and the context in which they may contribute to induce T cell anergy still need to be established. Our understanding of how T cell unresponsiveness is maintained has also changed. Uncoupled signaling from the TCR is not the only mechanism that keeps anergic T cells unresponsive. A series of epigenetic changes that include histone modifications and DNA methylation contribute to establish a stable repression of cytokine expression and play a key role in the maintenance of T cell anergy. Furthermore, we are now beginning to understand how the mechanisms that control T cell anergy and regulatory T cell function may integrate. The identification and characterization of the signaling networks that are responsible for the inhibition of TCR signaling and the suppression of cytokine transcription in anergic T cells would definitively offer new targets for the design of therapeutic approaches modulating tolerance to more effectively treat autoimmune diseases, allergy, cancer and graft rejection.
ACKNOWLEDGEMENTSThis work was supported by National Institutes of Health grants AI059738 and AI079363.
CONFLICT OF INTERESTThe authors declare no financial conflict of interes.