




El síndrome MELAS –miopatía, encefalopatía, acidosis láctica y episodios similares a ictus– es una citopatía mitocondrial relacionada con varias mutaciones del ADN mitocondrial, siendo la substitución A3243G en el gen tARNLeu la más frecuentemente asociada.
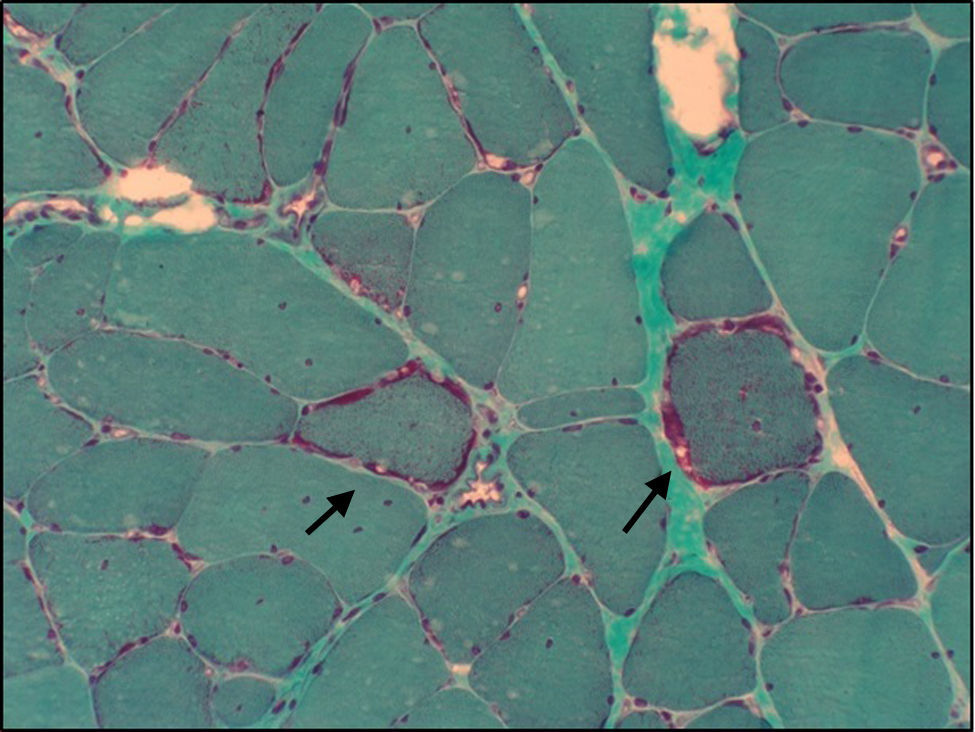
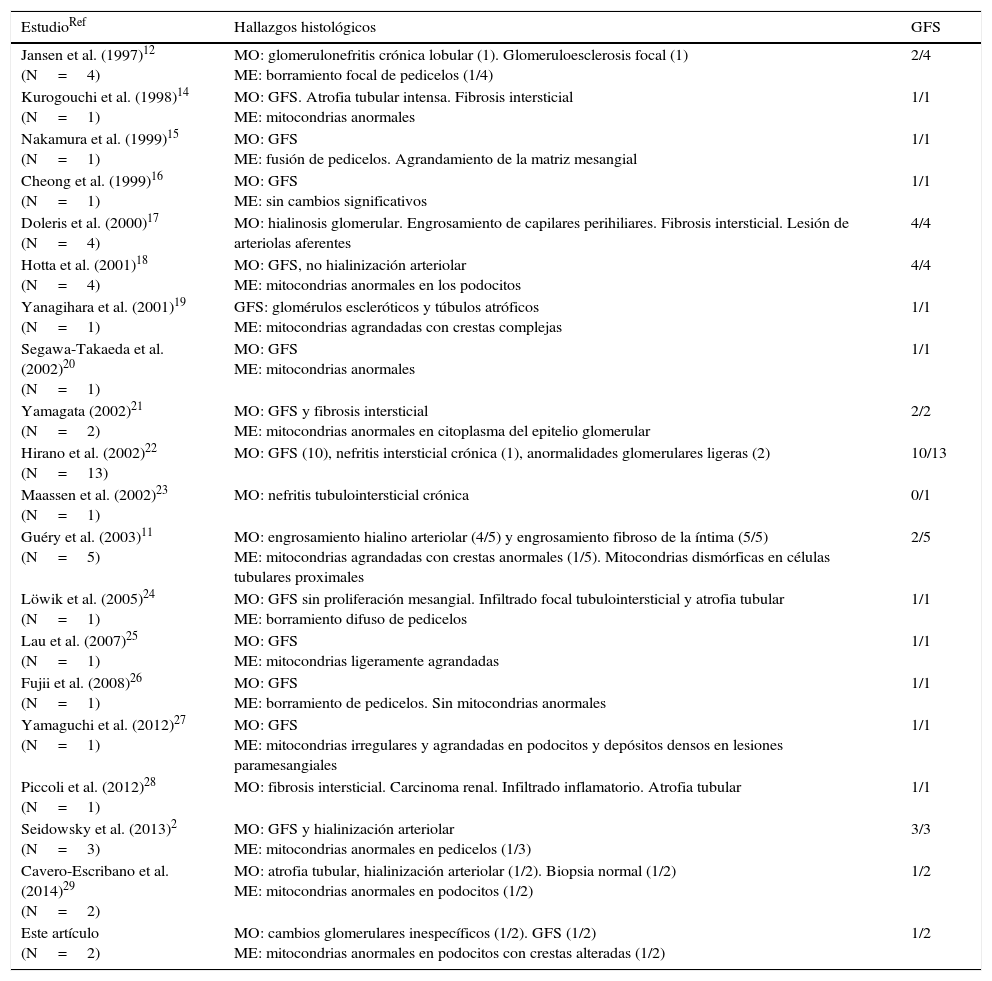
Pacientes y métodosAparte de su sintomatología habitual, los pacientes presentan historia de sordera neurosensorial y diabetes tipo 2 (DM2). Además, estudios recientes muestran que algunos pacientes tienen también afección renal, normalmente en forma de glomeruloesclerosis focal y segmentaria (GFS).
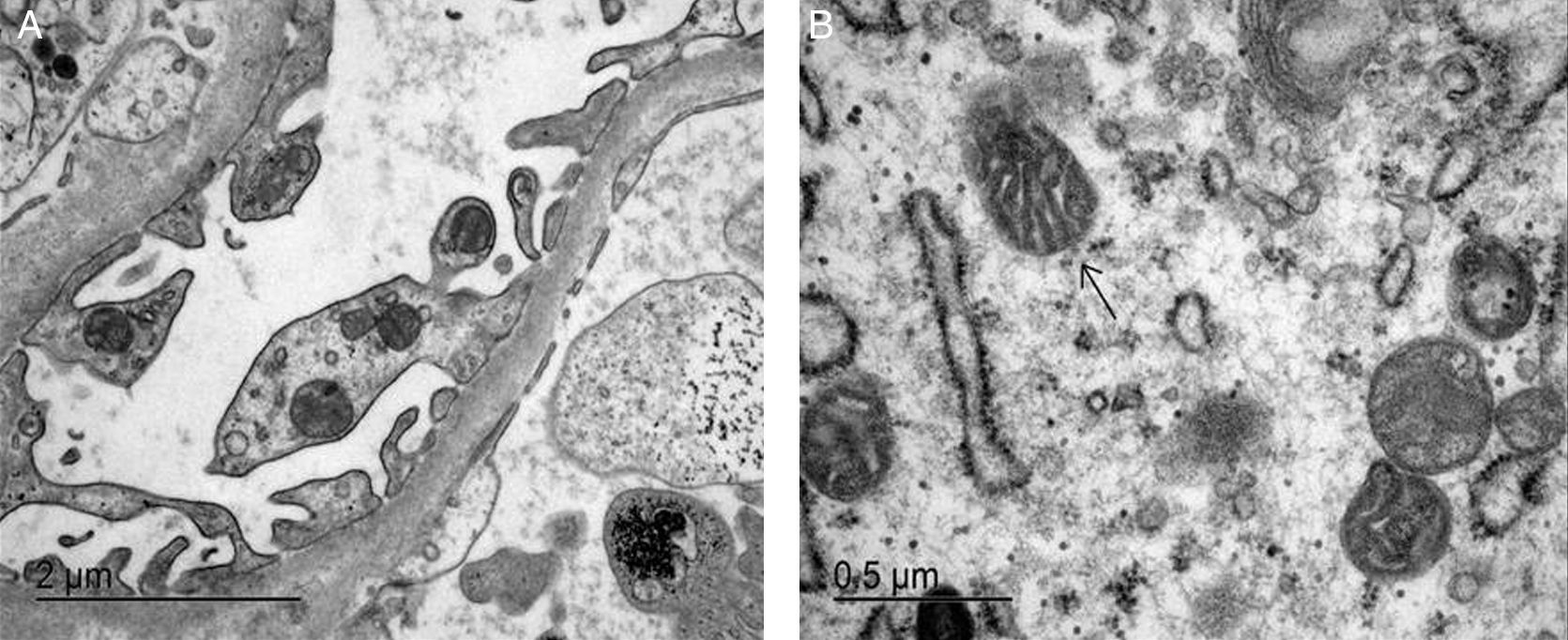
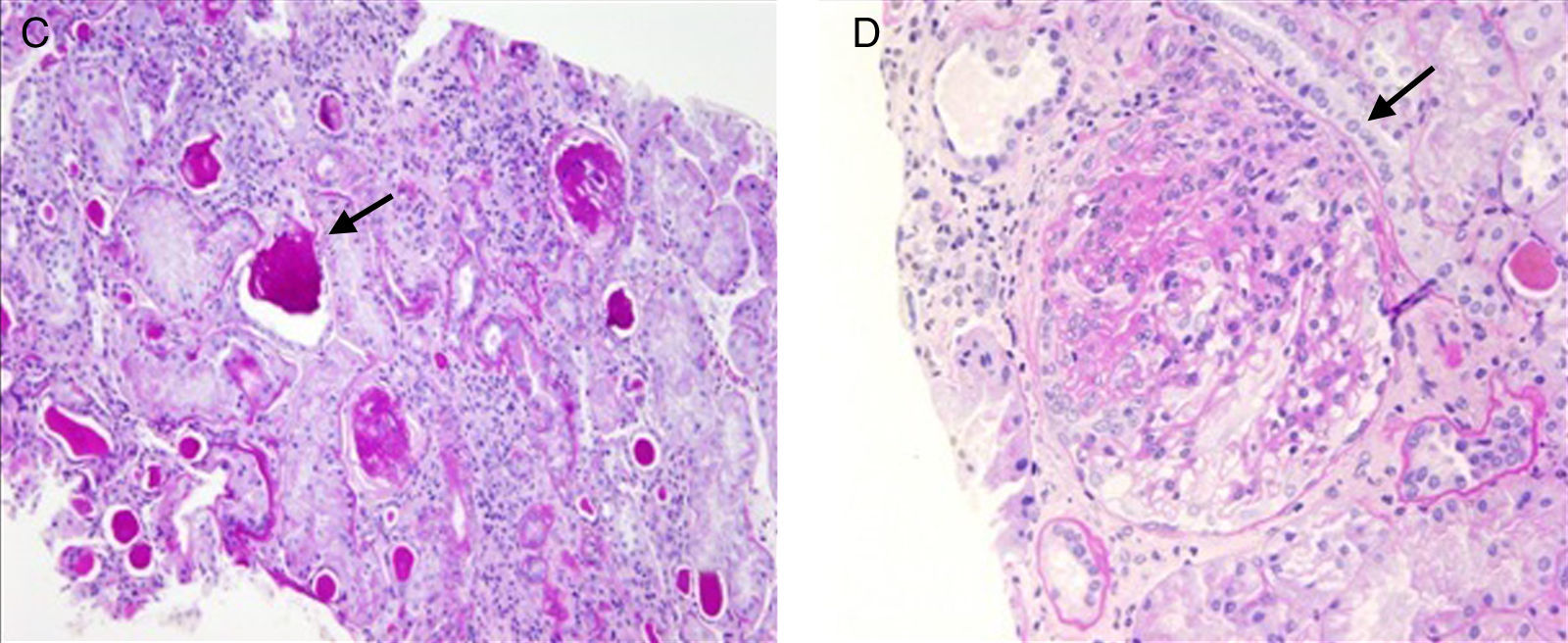
ResultadosEn este artículo se discute la afección renal de 2 pacientes no emparentados portadores de la mutación A3243G. Los 2 presentan sordera neurosensorial y DM2. Se realizó estudio anatomopatológico en ambos. Uno de ellos desarrolló proteinuria en rango nefrótico e insuficiencia renal terminal, con cambios de GFS en la biopsia, mientras que el otro presentaba proteinuria leve e insuficiencia renal, sin cambios histológicos reseñables en la microscopia óptica.
ConclusiónLa presencia de GFS u otra afección glomerular o tubular renal, acompañada de sordera neurosensorial y DM2, podría ser indicativa de la existencia de la mutación A3243G y estos hallazgos deberían propiciar un estudio genético y una evaluación de posible afección extrarrenal.
MELAS syndrome –myopathy, encephalopathy, lactic acidosis and stroke-like episodes– is a maternally-inherited mitochondrial cytopathy related to several mitochondrial DNA mutations, with the A3243G mutation in tRNALeu gene being the most frequent of them.
Patients and methodsApart from its typical symptomatology, patients usually exhibit a maternally-inherited history of neurosensory deafness and insulin-dependent type 2 diabetes mellitus (T2DM). Recent studies have shown that few patients carrying a A3243G mutation also suffer from renal dysfunction, usually in form of focal segmental glomerulosclerosis (FSGS).
ResultsIn this study we examine kidney involvement in 2 unrelated patients with a A3243G mutation by genetic testing. Both have a maternally-inherited neurosensory deafness and insulin-dependent T2DM. A renal biopsy was performed in both patients. One patient developed nephrotic proteinuria and renal insufficiency, with FSGS findings being observed in the kidney biopsy, whereas the other suffered from mild proteinuria and renal insufficiency, with non-specific glomerular changes.
ConclusionThe presence of FSGS or other kidney involvement accompanied by hereditary neurosensory deafness and T2DM could be suggestive of a A3243G tRNALeu mutation and should prompt a genetic testing and an evaluation of potential extrarenal involvement.
Artículo

Comprando el artículo el PDF del mismo podrá ser descargado
Precio 19,34 €
Comprar ahora
