




El síndrome MELAS –miopatía, encefalopatía, acidosis láctica y episodios similares a ictus– es una citopatía mitocondrial relacionada con varias mutaciones del ADN mitocondrial, siendo la substitución A3243G en el gen tARNLeu la más frecuentemente asociada.
Pacientes y métodosAparte de su sintomatología habitual, los pacientes presentan historia de sordera neurosensorial y diabetes tipo 2 (DM2). Además, estudios recientes muestran que algunos pacientes tienen también afección renal, normalmente en forma de glomeruloesclerosis focal y segmentaria (GFS).
ResultadosEn este artículo se discute la afección renal de 2 pacientes no emparentados portadores de la mutación A3243G. Los 2 presentan sordera neurosensorial y DM2. Se realizó estudio anatomopatológico en ambos. Uno de ellos desarrolló proteinuria en rango nefrótico e insuficiencia renal terminal, con cambios de GFS en la biopsia, mientras que el otro presentaba proteinuria leve e insuficiencia renal, sin cambios histológicos reseñables en la microscopia óptica.
ConclusiónLa presencia de GFS u otra afección glomerular o tubular renal, acompañada de sordera neurosensorial y DM2, podría ser indicativa de la existencia de la mutación A3243G y estos hallazgos deberían propiciar un estudio genético y una evaluación de posible afección extrarrenal.
MELAS syndrome –myopathy, encephalopathy, lactic acidosis and stroke-like episodes– is a maternally-inherited mitochondrial cytopathy related to several mitochondrial DNA mutations, with the A3243G mutation in tRNALeu gene being the most frequent of them.
Patients and methodsApart from its typical symptomatology, patients usually exhibit a maternally-inherited history of neurosensory deafness and insulin-dependent type 2 diabetes mellitus (T2DM). Recent studies have shown that few patients carrying a A3243G mutation also suffer from renal dysfunction, usually in form of focal segmental glomerulosclerosis (FSGS).
ResultsIn this study we examine kidney involvement in 2 unrelated patients with a A3243G mutation by genetic testing. Both have a maternally-inherited neurosensory deafness and insulin-dependent T2DM. A renal biopsy was performed in both patients. One patient developed nephrotic proteinuria and renal insufficiency, with FSGS findings being observed in the kidney biopsy, whereas the other suffered from mild proteinuria and renal insufficiency, with non-specific glomerular changes.
ConclusionThe presence of FSGS or other kidney involvement accompanied by hereditary neurosensory deafness and T2DM could be suggestive of a A3243G tRNALeu mutation and should prompt a genetic testing and an evaluation of potential extrarenal involvement.
El síndrome myopathy, encephalopathy, lactic acidosis and stroke-like (MELAS, «miopatía, encefalopatía, acidosis láctica y episodios similares a ictus») es una de las citopatías mitocondriales más frecuentes. Aunque la estructura y función del ADN mitocondrial (ADNmt) fueron descritas hace más de 40 años, las primeras mutaciones patógenas no se identificaron hasta 1988. Al menos 30 de ellas se han asociado con el síndrome MELAS, siendo la mutación A3243G tARNLeu la más frecuente (80%)1. Se han descrito una amplia variedad de citopatías mitocondriales diferentes asociadas a mutaciones puntuales que afectan a genes del ADNmt que codifican tRNA2.
Aunque es poco frecuente, la afección renal en el espectro del síndrome MELAS se ha descrito en diversas series de casos. Parece que la forma más frecuente de presentación es la glomeruloesclerosis focal y segmentaria (GFS), aunque se han descrito otras lesiones. En este artículo se describe la afección renal y las complicaciones de 2 pacientes no emparentados, portadores de la mutación A3243G, detectada mediante pruebas genéticas.
Caso 1Mujer de 60 años, sin alergias ni hábitos tóxicos, que fue remitida al Servicio de Nefrología por el hallazgo incidental en un análisis de sangre sistemático de una creatinina sérica de 1,49mg/dl.
Como antecedentes, destacan una diabetes mellitus tipo 2 (DM2) diagnosticada a la edad de 36 años, que recibió tratamiento con antidiabéticos orales durante 8 años, progresando después a tratamiento con insulina. Los controles glucémicos eran correctos, con niveles de HbA1c entre 5,4 y 6,5%. El fondo de ojo no había revelado signos de retinopatía diabética y no había evidencia de ninguna afección microvascular ni de órganos diana. También presentaba una sordera neurosensorial de etiología desconocida, diagnosticada antes de los 25 años. Ocho años antes, se le diagnosticó un melanoma localizado en su rodilla derecha, estando actualmente libre de enfermedad. La paciente negó historia familiar de DM2 o sordera neurosensorial. En el examen, su peso era de 45kg y su altura de 152cm, con un IMC de 19kg/m2. Su presión arterial era de 111/72mmHg, la frecuencia cardíaca de 56lpm y el examen físico por aparatos, sin hallazgos significativos. Su creatinina en suero fue de 1,5mg/dl, con un filtrado glomerular estimado de 33ml/min/m2 y un BUN de 39mg/dl; el equilibrio venoso no mostró acidosis metabólica y los electrólitos séricos fueron normales. Hemograma, pruebas de función hepática, lípidos séricos, creatinina cinasa (CK) y ácido úrico en suero estaban dentro de la normalidad. Los análisis de orina no mostraron hematuria ni proteinuria. La ecografía renal mostró una reducción cortical leve y varios pequeños quistes corticales. A pesar de que la paciente no tenía historial familiar de sordera neurosensorial o DM2, y que no mostraba síntomas evidentes de citopatía mitocondrial (es decir, acidosis láctica, intolerancia al ejercicio o miopatía o síntomas neurológicos), se sospechó una enfermedad mitocondrial. Una prueba genética en la orina confirmó una mutación A3243G con un 29% de heteroplasmia. En el seguimiento, los niveles de creatinina se mantuvieron estables y el sedimento de orina y la proteinuria permanecieron normales. Aunque se propuso una biopsia renal, la paciente la rechazó.
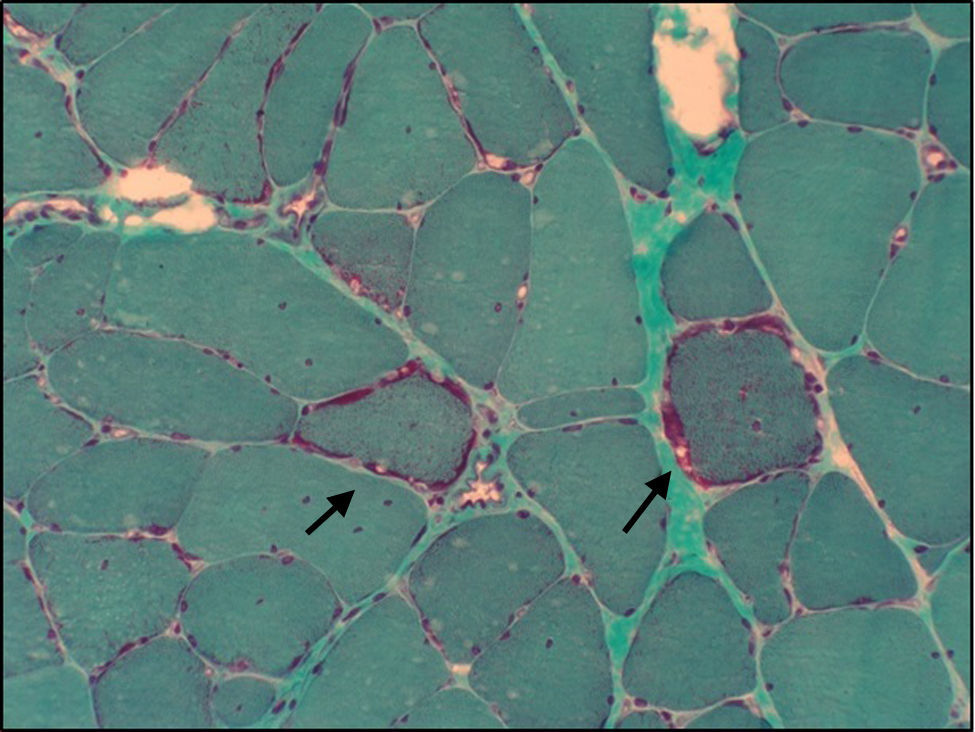
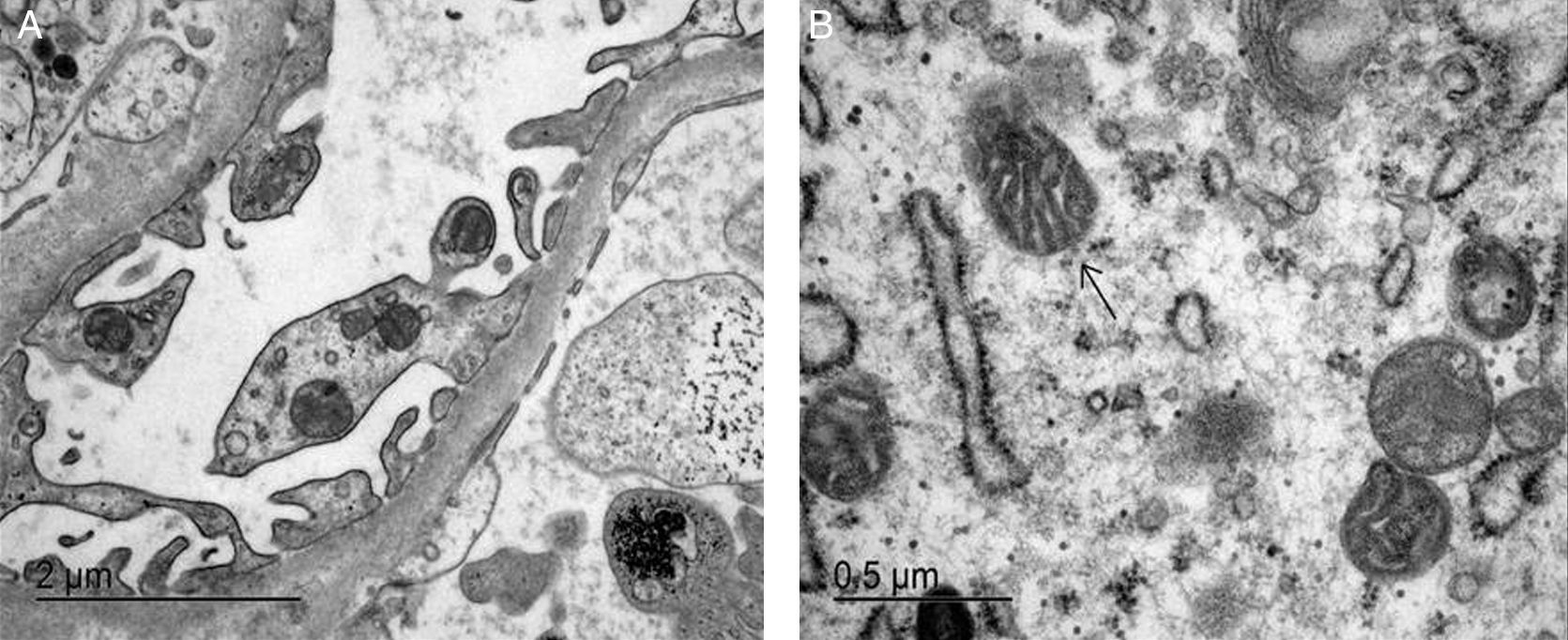
Tres años después de la primera visita, la paciente aquejó un aumento de cansancio y debilidad generalizada e intolerancia al ejercicio. Los análisis de sangre mostraron CK 2.819U/l, LDH 1.571U/l y aldolasa 31,7U/l (valores normales entre 0,3-6U/l). En el examen físico se halló dificultad en el habla y ptosis palpebral bilateral, junto con una leve pérdida generalizada de la fuerza muscular (4/5). Su función cognitiva era normal. Se sometió a una biopsia muscular, que mostró signos inequívocos de miopatía mitocondrial (fig. 1). En analíticas posteriores, CK, LDH y aldolasa se normalizaron, a pesar de la persistencia de los síntomas. No se consideró ningún tratamiento específico, excepto terapia física y ejercicio aeróbico. Posteriormente, se observó un leve aumento de la creatinina sérica (a 1,6mg/dl) y se realizó una biopsia renal con el fin de profundizar en el estudio de su insuficiencia renal incipiente en el contexto de un síndrome MELAS. Se hallaron cambios glomerulares e intersticiales inespecíficos en la microscopia óptica. Sin embargo, el examen ultraestructural con microscopia electrónica reveló un borramiento focal de los pedicelos de los podocitos y mitocondrias anormales, muchas de ellas dentro de los pedicelos (fig. 2).
. Se observan varias fibras típicas «rojas rasgadas» (flechas negras), así como núcleos en el interior de algunas fibras y variabilidad en el tamaño de estas. Todo ello es compatible con miopatía mitocondrial.")
Biopsia muscular de la paciente 1 (tinción modificada de tricrómico de Gomori para tejido congelado). Se observan varias fibras típicas «rojas rasgadas» (flechas negras), así como núcleos en el interior de algunas fibras y variabilidad en el tamaño de estas. Todo ello es compatible con miopatía mitocondrial.

Imagen de microscopia electrónica extraída de la biopsia de la paciente 1. A. Fusión focal de pedicelos, con mitocondrias anormales, hallazgo específico de la mutación A3243G. B. La flecha negra apunta a una mitocondria anormal hallada en el citoplasma del podocito, con cierto desordenamiento de las crestas.
Cinco años después de la primera visita, la paciente desarrolló un cuadro de disfagia y alteraciones de la marcha. Aunque en ese momento se detectó una albuminuria leve de 300mg/g de creatinina, la función renal se mantuvo estable.
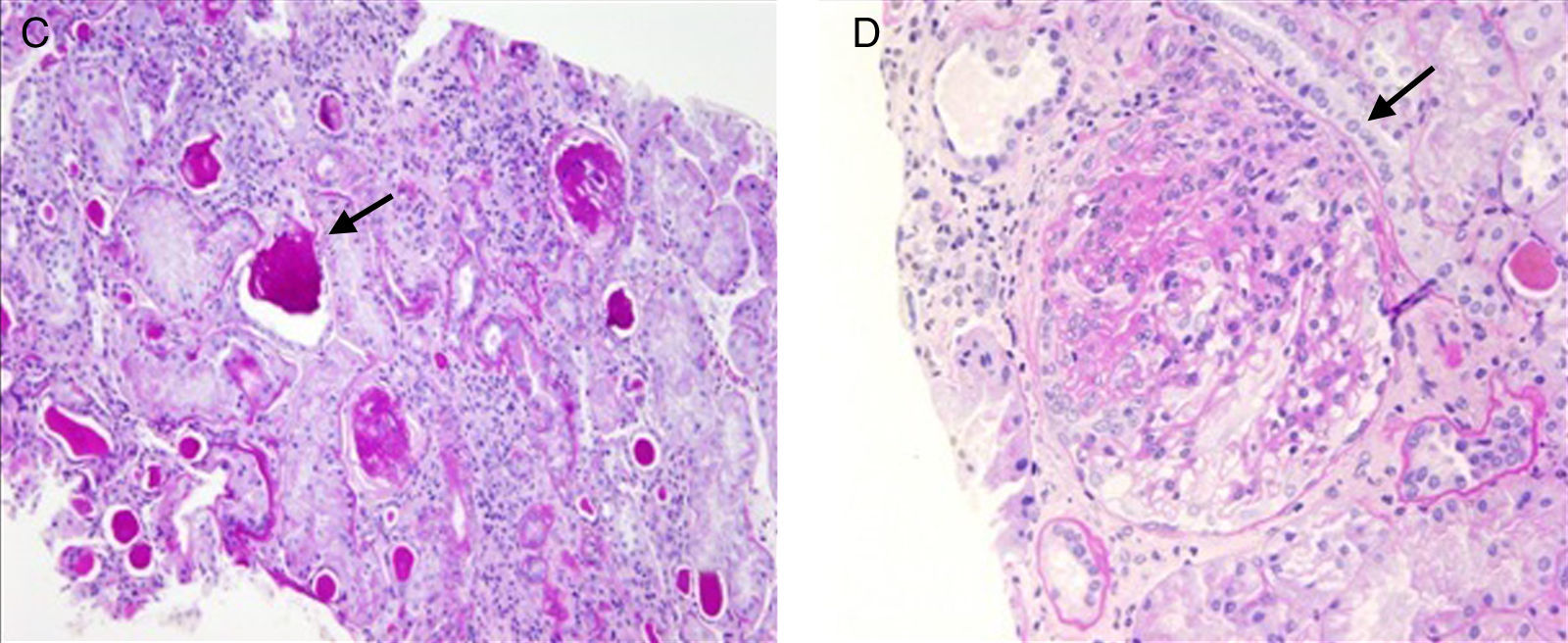
Caso 2Un paciente varón de 30 años de edad, sin alergias ni hábitos tóxicos, es visitado en el Servicio de Nefrología por hallazgo de proteinuria de 3,15g en orina de 24h y creatinina sérica de 1,5mg/dl. Destaca el antecedente de madre fallecida por coma diabético. No tenía antecedentes de sordera neurosensorial. Como antecedentes patológicos destaca, a la edad de 12 años, un episodio de rabdomiolisis con insuficiencia renal aguda después de pasar 8h caminando. Pasó 30 días en el hospital y recibió 20 sesiones de hemodiálisis. La función renal se recuperó totalmente después del episodio. Su peso era de 68kg y su altura era de 170cm; IMC de 23kg/m2. El examen físico fue normal, excepto hipertensión leve, controlada correctamente con inhibidores de la ECA, y sordera neurosensorial unilateral leve diagnosticada mediante pruebas audiométricas, que se mantuvo estable. No se detectó hematuria. La cantidad de proteinuria se redujo después del tratamiento con inhibidores de la ECA a niveles entre 1,5 y 2,5g/24h, sin hipoalbuminemia, edema o hiperlipidemia. Debido al grado de proteinuria, el paciente fue sometido a biopsia renal, que puso de manifiesto hallazgos típicos de GSF con presencia de C3 e IgM+++ (fig. 3). En el seguimiento, el paciente desarrolló progresivamente intolerancia al ejercicio, debilidad en las extremidades y cansancio, junto con enzimas musculares anormales, que inicialmente aumentaron ligeramente (CK 3.47U/l; LDH 506U/l), y luego de forma progresiva en el tiempo, a niveles de 2.308U/l para CK y de 2.324U/l para LDH, con empeoramiento de la sintomatología. El estudio electromiográfico no halló alteraciones, pero una biopsia muscular mostró varias células musculares tipo ragged red o «rojas rasgadas», algunas con depósitos de lípidos, muy indicativas de miopatía mitocondrial. Un análisis genético en orina manifestó una mutación A3243G con un 32% de heteroplasmia. Con el fin de estudiar más a fondo la afectación de órganos, se le realizó un ecocardiograma, que no mostró alteraciones morfológicas, con una FEVI del 54%. Una resonancia magnética craneal mostró una importante atrofia cerebral, con cavidades ventriculares agrandadas.
. Se observan varios glomérulos escleróticos (A, flecha negra). También se pueden observar cambios típicos de GFS en estos glomérulos. En la mitad superior se observa colapso segmentario con esclerosis (B, flecha negra).")
En ese momento, el paciente fue diagnosticado de DM2 con hiperinsulinismo, con una HbA1c de 8,8%, sin retinopatía. En un primer momento respondió adecuadamente a antidiabéticos orales. Sin embargo, el control glucémico resultó inadecuado durante el seguimiento, con valores de HbA1c que alcanzaron el 11,1%, momento en el cual se introdujo tratamiento con insulina.
Durante el seguimiento, la proteinuria fue apenas controlada, con valores entre 2 y 5g/24h a pesar del control óptimo de la presión arterial. La función renal se redujo progresivamente, llegando a enfermedad renal terminal 15 años más tarde, momento en que se inició hemodiálisis.
DiscusiónEn el espectro de la mutación A3243G, la afección renal es rara. La sintomatología común relacionada con esta mutación incluye: intolerancia al ejercicio (93%)1, pérdida de audición (44-77%)1,2, miopatía y debilidad de las extremidades (37-53%)2,3, DM2 (12-33%)1–3, inicio sintomatológico en la edad adulta (40%)3 e historia familiar de MELAS en menos del 20%3. La prevalencia de esta mutación en pacientes con enfermedad renal se ha reportado en numerosas ocasiones, aunque con resultados controvertidos4,5. Más estudios que incluyan más pacientes de diferentes grupos étnicos serían necesarios para establecer de manera precisa la incidencia y la prevalencia de dicha mutación en pacientes con enfermedad renal crónica. En la población general, la presencia de la mutación A3243G se ha estimado entre 16,3 y 236/100.0006–9. Hoy en día se acepta que las citopatías mitocondriales son la enfermedad metabólica más frecuente en humanos1.
La manifestación renal más frecuente en citopatías mitocondriales en general, y en la edad pediátrica en particular, es el síndrome de Toni-Debré-Fanconi, una disfunción del túbulo proximal caracterizada por un deterioro de la reabsorción de glucosa, aminoácidos, fosfato y bicarbonato, lo que lleva a una posterior glucosuria, aminoaciduria, hiperfosfaturia y acidosis tubular proximal10. Ninguno de los pacientes estudiados mostró manifestaciones de síndrome de Fanconi.
En el síndrome MELAS, la anomalía renal más frecuente encontrada en la edad adulta es la GFS, una glomerulonefritis no proliferativa caracterizada por esclerosis glomerular parcial, que afecta solo a algunos glomérulos (focal), y entre los afectados, solo algunos capilares muestran cambios patológicos (segmentaria)2. En nuestra serie de casos, ambos pacientes fueron biopsiados. La paciente 1 mostró cambios inespecíficos en la microscopia óptica, y presencia de mitocondrias anormales dentro de los podocitos en la microscopia electrónica. El hallazgo de mitocondrias dentro de los pedicelos de los podocitos se propone como patognomónico de MELAS11. El paciente 2 mostró cambios de GFS típicos en la microscopia óptica. En este paciente no se realizaron técnicas de microscopia electrónica.
Ambos pacientes presentaban DM2, diagnosticada a temprana edad. Ninguno de los pacientes presentaba sobrepeso. La presencia de DM2 en pacientes atípicos, especialmente con antecedentes familiares, es muy indicativa de una causa subyacente. Esta alta prevalencia de DM2 en pacientes con afección renal en el síndrome MELAS ya se ha reportado con anterioridad11, lo que evidencia una posible relación fisiopatológica. Aunque la paciente 1 fue diagnosticada de DM2 años antes, no se observaron signos de nefropatía diabética en la biopsia renal. En el paciente 2, la DM2 se diagnosticó después de la biopsia renal, pero su falta de control metabólico posiblemente influyó en el rápido empeoramiento de la función renal.
Ambos pacientes también presentaban algún grado de sordera neurosensorial. La pérdida de audición añadida a la insuficiencia renal es altamente indicativa de síndrome de Alport, y el diagnóstico diferencial entre esta entidad y una citopatía mitocondrial con afección renal se debe tener siempre en cuenta2.
Cabe destacar que ninguno de los pacientes ha presentado episodios similares a ictus u otra sintomatología neurológica aparte de la sordera, aunque en el paciente 2 se demostró mediante pruebas de neuroimagen una atrofia cerebral importante con aumento difuso del volumen ventricular, que podrían corresponderse en parte a cambios secundarios a la afectación mitocondrial en el SNC.
La paciente 1 solo presentaba DM2, pérdida de la audición y miopatía, además de afección renal no diabética. Este subtipo de presentación clínica se caracteriza principalmente por maternally inherited diabetes and deafness (MIDD, «diabetes y sordera de herencia materna») y ausencia de otros síntomas «clásicos» del síndrome MELAS, como acidosis láctica o encefalopatía12. A pesar de que la paciente 1 no tenía historia familiar de diabetes y sordera, la presentación clínica es más indicativa de MIDD que de un síndrome MELAS clásico. Un estudio de cohortes prospectivo de 54 pacientes con MIDD, portadores de la mutación A3243G, mostró una disociación significativa entre la afección renal y la retinopatía diabética (28 frente al 8%), lo que evidencia que la implicación específicamente renal es secundaria a la enfermedad mitocondrial13. Las diferencias fisiopatológicas entre el síndrome MELAS clásico y el MIDD se desconocen, pero probablemente están en relación con la heteroplasmia y con fenómenos de deriva génica.
Como hemos visto, también existe variación de la presentación de la sintomatología renal. La paciente 1 solo mostró una elevación ligera de los valores de creatinina con albuminuria leve esporádica y una reducción moderada de la tasa de filtración glomerular, mientras que el caso 2 presentó en la infancia rabdomiolisis inducida por el ejercicio y, secundariamente, una insuficiencia renal aguda. La biopsia renal de la paciente 1 solo mostró cambios inespecíficos en el microscopio óptico. En el microscopio electrónico se observaron mitocondrias anormales en los podocitos. Se puede especular que estas mitocondrias anormales que se encuentran en los podocitos podrían ser un signo de posible daño celular futuro, que pudiera conducir a esta paciente al desarrollo de una GSF establecida o bien a un síndrome de Fanconi.
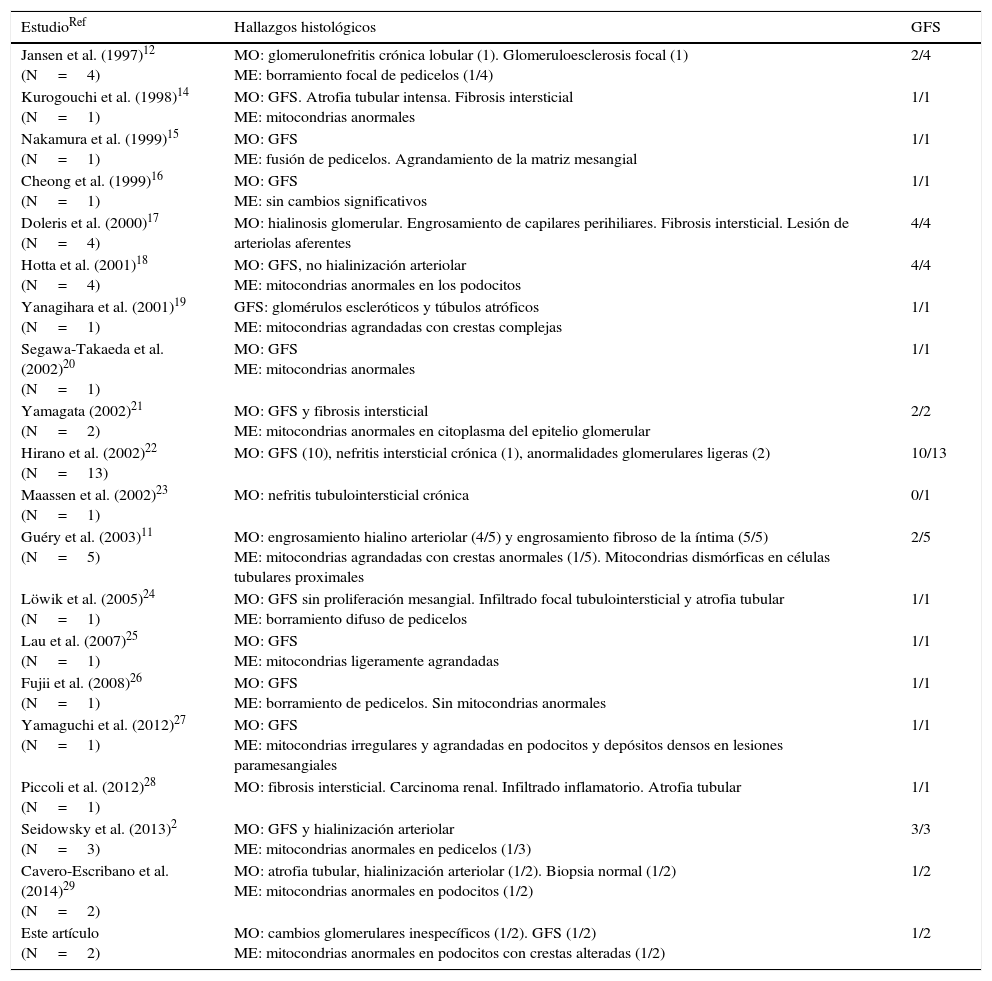
La tabla 1 muestra los hallazgos patológicos en los casos publicados de síndrome MELAS (mutación ADNmt A3243G) con afección renal. De los 50 pacientes examinados anteriormente, 39 (78%) muestran características histológicas de la GFS. En observaciones con microscopia electrónica, parece ser significativa la aparición de mitocondrias anormales en los pedicelos. El mecanismo por el cual se produce daño selectivo de los pedicelos de los podocitos no está aclarado, pero podría tener relación con la presencia de mitocondrias anormales, con las vías apoptóticas alteradas y/o con la producción de radicales libres.
Resumen de los hallazgos patológicos en pacientes con mutación en el ADN mitocondrial y afección renal reportados en la literatura médica
| EstudioRef | Hallazgos histológicos | GFS |
|---|---|---|
| Jansen et al. (1997)12 (N=4) | MO: glomerulonefritis crónica lobular (1). Glomeruloesclerosis focal (1) ME: borramiento focal de pedicelos (1/4) | 2/4 |
| Kurogouchi et al. (1998)14 (N=1) | MO: GFS. Atrofia tubular intensa. Fibrosis intersticial ME: mitocondrias anormales | 1/1 |
| Nakamura et al. (1999)15 (N=1) | MO: GFS ME: fusión de pedicelos. Agrandamiento de la matriz mesangial | 1/1 |
| Cheong et al. (1999)16 (N=1) | MO: GFS ME: sin cambios significativos | 1/1 |
| Doleris et al. (2000)17 (N=4) | MO: hialinosis glomerular. Engrosamiento de capilares perihiliares. Fibrosis intersticial. Lesión de arteriolas aferentes | 4/4 |
| Hotta et al. (2001)18 (N=4) | MO: GFS, no hialinización arteriolar ME: mitocondrias anormales en los podocitos | 4/4 |
| Yanagihara et al. (2001)19 (N=1) | GFS: glomérulos escleróticos y túbulos atróficos ME: mitocondrias agrandadas con crestas complejas | 1/1 |
| Segawa-Takaeda et al. (2002)20 (N=1) | MO: GFS ME: mitocondrias anormales | 1/1 |
| Yamagata (2002)21 (N=2) | MO: GFS y fibrosis intersticial ME: mitocondrias anormales en citoplasma del epitelio glomerular | 2/2 |
| Hirano et al. (2002)22 (N=13) | MO: GFS (10), nefritis intersticial crónica (1), anormalidades glomerulares ligeras (2) | 10/13 |
| Maassen et al. (2002)23 (N=1) | MO: nefritis tubulointersticial crónica | 0/1 |
| Guéry et al. (2003)11 (N=5) | MO: engrosamiento hialino arteriolar (4/5) y engrosamiento fibroso de la íntima (5/5) ME: mitocondrias agrandadas con crestas anormales (1/5). Mitocondrias dismórficas en células tubulares proximales | 2/5 |
| Löwik et al. (2005)24 (N=1) | MO: GFS sin proliferación mesangial. Infiltrado focal tubulointersticial y atrofia tubular ME: borramiento difuso de pedicelos | 1/1 |
| Lau et al. (2007)25 (N=1) | MO: GFS ME: mitocondrias ligeramente agrandadas | 1/1 |
| Fujii et al. (2008)26 (N=1) | MO: GFS ME: borramiento de pedicelos. Sin mitocondrias anormales | 1/1 |
| Yamaguchi et al. (2012)27 (N=1) | MO: GFS ME: mitocondrias irregulares y agrandadas en podocitos y depósitos densos en lesiones paramesangiales | 1/1 |
| Piccoli et al. (2012)28 (N=1) | MO: fibrosis intersticial. Carcinoma renal. Infiltrado inflamatorio. Atrofia tubular | 1/1 |
| Seidowsky et al. (2013)2 (N=3) | MO: GFS y hialinización arteriolar ME: mitocondrias anormales en pedicelos (1/3) | 3/3 |
| Cavero-Escribano et al. (2014)29 (N=2) | MO: atrofia tubular, hialinización arteriolar (1/2). Biopsia normal (1/2) ME: mitocondrias anormales en podocitos (1/2) | 1/2 |
| Este artículo (N=2) | MO: cambios glomerulares inespecíficos (1/2). GFS (1/2) ME: mitocondrias anormales en podocitos con crestas alteradas (1/2) | 1/2 |
GFS: glomeruloesclerosis focal y segmentaria; ME: microscopia electrónica; MO: microscopia óptica.
En conclusión, la afectación renal es variable en pacientes con la mutación A3243G. La presencia de GFS u otra afección renal crónica, acompañada de sordera neurosensorial hereditaria y DM2, puede indicar la presencia de la mutación A3243G tARNLeu y, por lo tanto, estos hallazgos deben conllevar la realización de pruebas genéticas y una extensa evaluación de la potencial afectación extrarrenal de dicha enfermedad.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
