





La hemoglobina es un tetrámero constituido por dos cadenas α y dos cadenas β. Los dos genes estructurales que codifican la cadena α se encuentran localizados en el brazo corto del cromosoma 16. Los individuos normales tienen 4 genes α (αα/αα). Las α talasemias se producen generalmente por la deleción de uno, dos, tres o cuatro de los genes α. La deleción de ambos genes α dentro del mismo cromosoma (α° talasemia) se observa en individuos del área mediterránea y en el sudeste asiático.
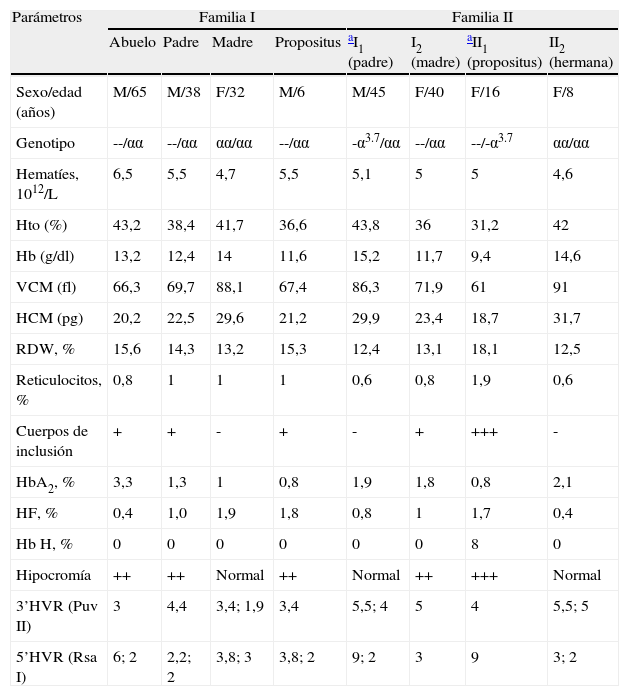
Material y métodoSe estudian dos familias naturales de Madrid con anemia microcítica e hipocroma. El ADN extraído de los leucocitos de sangre periférica se digiere con diferentes enzimas de restricción e hibridación con sondas del cluster de genes α. Los extremos de la deleción se han caracterizado combinando las técnicas de Southern blot, reacción en cadena de la polimerasa (PCR) e hibridación in situ fluorescente (FISH).
ResultadosEn este trabajo presentamos dos nuevas mutaciones de α° talasemia en dos familias españolas, no descritas previamente en la bibliografía. La mutación (--ED) presenta una deleción de alrededor de 80kb con el punto de rotura 5’ en la coordenada +100 (± 3kb), mientras que el extremo 3’ HVR se sitúa en la coordenada 178±750bp. La segunda mutación (--GP) es más extensa, con pérdida de 145kb, situándose la deleción en la región 5’, entre las coordenadas 34 y 37, respetando el telómero. En la región centromérica la rotura se sitúa también en la coordenada 178±1,4kb. En ambas mutaciones se pierden los dos genes α, el gen θ y la región reguladora HS40.
ConclusionesLa exacta identificación de estas mutaciones es esencial para determinar la función de los genes α en caso de consejo genético.
The two structural genes encoding the human α-globin chains are located on the short arm of chromosome 16. Normal individuals have four genes α (αα/αα). α-thalassemias are usually produced by the deletion of one, two, three, or four α genes. Deletion of both α genes within the same chromosome (α° thalassemia) is commonly observed in individuals from the Mediterranean basin and Southeast Asia.
Material and methodsWe study two natural families of Madrid with microcytic hypochromic anemia. The DNA extracted from peripheral blood leukocytes was digested with different restriction enzymes and hybridization with probes of gene cluster α. The ends of the deletion were characterized by combining the techniques of Southern blot, PCR and FISH.
ResultsWe present two new mutations of α° thalassemia in two Spanish families, not previously described in the literature. The deletion (--ED) is ∼80kb with the point of bread 5’ in the coordinate +100 (± 3kb), whereas the end 3’HVR places in the coordinate 178±750bp. The second deletion (--GP) is more extensive, with loss of 145kb, placing the deletion in the end 5’ between the coordinates 34 and 37, respecting therefore the telomere. In the centromeric region the point of break places as the previous one in the coordinate 178±1.4bp.
ConclusionsIn both mutations both alpha genes were deleted, the gene θ and the region HS40. The exact identification of these deletions is essential to determine the function of the genes α with a view to a possible genetic diagnosis.
Artículo

Comprando el artículo el PDF del mismo podrá ser descargado
Precio 19,34 €
Comprar ahora
