





La hemoglobina es un tetrámero constituido por dos cadenas α y dos cadenas β. Los dos genes estructurales que codifican la cadena α se encuentran localizados en el brazo corto del cromosoma 16. Los individuos normales tienen 4 genes α (αα/αα). Las α talasemias se producen generalmente por la deleción de uno, dos, tres o cuatro de los genes α. La deleción de ambos genes α dentro del mismo cromosoma (α° talasemia) se observa en individuos del área mediterránea y en el sudeste asiático.
Material y métodoSe estudian dos familias naturales de Madrid con anemia microcítica e hipocroma. El ADN extraído de los leucocitos de sangre periférica se digiere con diferentes enzimas de restricción e hibridación con sondas del cluster de genes α. Los extremos de la deleción se han caracterizado combinando las técnicas de Southern blot, reacción en cadena de la polimerasa (PCR) e hibridación in situ fluorescente (FISH).
ResultadosEn este trabajo presentamos dos nuevas mutaciones de α° talasemia en dos familias españolas, no descritas previamente en la bibliografía. La mutación (--ED) presenta una deleción de alrededor de 80kb con el punto de rotura 5’ en la coordenada +100 (± 3kb), mientras que el extremo 3’ HVR se sitúa en la coordenada 178±750bp. La segunda mutación (--GP) es más extensa, con pérdida de 145kb, situándose la deleción en la región 5’, entre las coordenadas 34 y 37, respetando el telómero. En la región centromérica la rotura se sitúa también en la coordenada 178±1,4kb. En ambas mutaciones se pierden los dos genes α, el gen θ y la región reguladora HS40.
ConclusionesLa exacta identificación de estas mutaciones es esencial para determinar la función de los genes α en caso de consejo genético.
The two structural genes encoding the human α-globin chains are located on the short arm of chromosome 16. Normal individuals have four genes α (αα/αα). α-thalassemias are usually produced by the deletion of one, two, three, or four α genes. Deletion of both α genes within the same chromosome (α° thalassemia) is commonly observed in individuals from the Mediterranean basin and Southeast Asia.
Material and methodsWe study two natural families of Madrid with microcytic hypochromic anemia. The DNA extracted from peripheral blood leukocytes was digested with different restriction enzymes and hybridization with probes of gene cluster α. The ends of the deletion were characterized by combining the techniques of Southern blot, PCR and FISH.
ResultsWe present two new mutations of α° thalassemia in two Spanish families, not previously described in the literature. The deletion (--ED) is ∼80kb with the point of bread 5’ in the coordinate +100 (± 3kb), whereas the end 3’HVR places in the coordinate 178±750bp. The second deletion (--GP) is more extensive, with loss of 145kb, placing the deletion in the end 5’ between the coordinates 34 and 37, respecting therefore the telomere. In the centromeric region the point of break places as the previous one in the coordinate 178±1.4bp.
ConclusionsIn both mutations both alpha genes were deleted, the gene θ and the region HS40. The exact identification of these deletions is essential to determine the function of the genes α with a view to a possible genetic diagnosis.
La hemoglobina (Hb) es un tetrámero constituido por dos cadenas α y dos cadenas β. Los dos genes estructurales α se encuentran localizados en el brazo corto del cromosoma 16. La talasemia α se produce por la deleción de uno, dos, tres o los cuatro genes α1. Un porcentaje pequeño de α talasemia se produce por mutaciones puntuales2.
La pérdida de 2 genes α en el mismo cromosoma produce α° talasemia. Se han descrito alrededor de 50 variantes de α° talasemia; algunas afectan a una sola familia3,4, mientras que otras son relativamente frecuentes en una determinada área geográfica1.
Recientemente hemos estudiado 2 familias con dos nuevas mutaciones de α° talasemia, una de ellas con 3 miembros afectados. En la segunda familia, el propositus presenta enfermedad de la Hb H y su madre α° talasemia, siendo ambos portadores de una nueva mutación de α° talasemia.
Material y métodoEn la familia 1 el propositus de 6 años es natural de Madrid, así como toda su familia. Consulta por moderada anemia microcítica e hipocroma (tabla 1).
En la familia 2 se trata de una paciente de 16 años de edad, también natural de Madrid, que presenta anemia hemolítica microcítica e hipocroma. Su madre tiene α° talasemia, y su padre α+ talasemia heterocigota (αα/-α3.7). Una hermana es normal (tabla 1).
El ADN de los leucocitos de la sangre periférica se extrae con fenol cloroformo y se digiere con diferentes enzimas de restricción, que se especifican en la tabla 1, y se hibridan con las siguientes sondas: fragmento ζ 1,8kb Sac I, obtenido de PRB ζ, fragmento α de 1,5kb Pst del p recombinante PRBα, fragmento 3’HVR 4,0kb Hinf I, obtenido del plásmido pSEA y fragmento 5’HVR 0,8kb Eco RI-Hind III obtenido del p 5’HVR.
Para el estudio de las mutaciones desarrollamos una línea celular híbrida humano/murina por fusión de linfocitos humanos transformados por el virus de Epstein Barr y células MEL de ratón, que retienen únicamente el cromosoma anormal humano sometiendo las células a metotrexato, que inhibe la enzima DHER, por lo que el gen humano del cromosoma 16 APRT se hace imprescindible.
Los extremos de la deleción se han caracterizado combinando las técnicas de Southern blot, reacción en cadena de la polimerasa (PCR) e hibridación fluorescente in situ (FISH)5. También se ha empleado la técnica de Multiplex Ligation dependent Probe Amplification (MLPA) con el objeto de delimitar los extremos 5’ y 3’ de la deleción según Harteveld et al6.
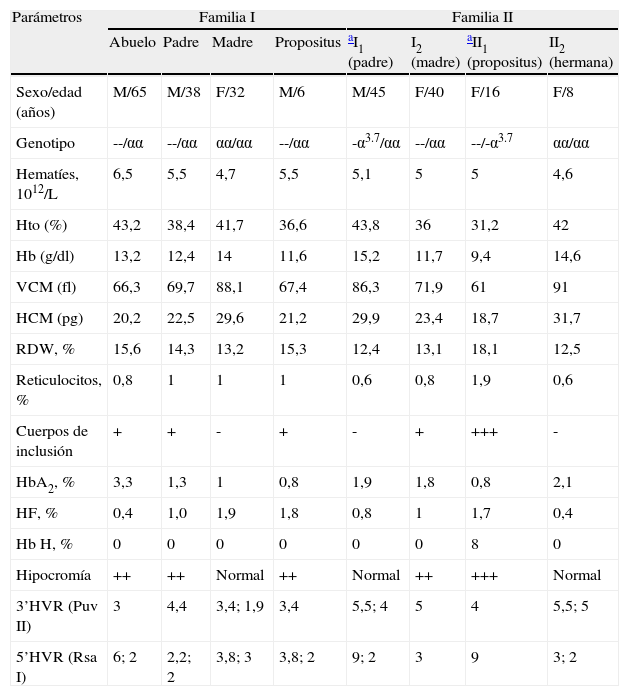
ResultadosLos datos hematológicos de los padres y el niño se observan en la tabla 1. El abuelo, padre y nieto son portadores de la mutación α°.
Con Bam HI, Bgl II, Eco RI, Hind III y Hpa I (sondas α y ζ) sólo se observan fragmentos normales debidos al cromosoma normal.
Los estudios de los 4 miembros de la familia con Pvu II (sonda 3’HVR) y Rsa I (sonda 5’HVR) se especifican al final de la tabla 1. Con 3’HVR sólo se obtiene una banda, mientras que en el extremo 5’HVR los dos alelos están presentes.
En la segunda familia la madre es portadora de una α° talasemia en un alelo y el padre presenta la mutación - α3.7.
El propositus tiene enfermedad de la Hb H (II1). Sus datos hematológicos y el estudio de ADN se especifican en la tabla 1. El estudio de la mutación α° con varias enzimas de restricción y su hibridación con varias sondas sólo demuestra bandas normales del cromosoma normal. El estudio con las enzimas Pvu (3’HVR) y Rsa I (5’HVR) sólo demuestra una sola banda, indicando que la deleción afecta a la totalidad del cluster α. En I1 y II1 se observan los fragmentos anormales correspondientes a la mutación –α3.7.
Con el objeto de determinar la extensión precisa de ambas deleciones, se estudiaron ambas familias en la línea celular híbrida con FISH. En la primera familia el estudio por PCR en el extremo telomérico mostró que la deleción se establecía entre los plásmidos 100580 a 103385, con una diferencia del punto de corte de aproximadamente 3 Kb. La combinación de Southern blot y PCR permiten determinar la deleción en el extremo 3’HVR, entre los plásmidos 177265 y 178773, con una diferencia en el punto de corte de 0,75kb (tabla 1). Por lo tanto, la deleción α (--ED) es de aproximadamente 80kb (fig. 1).
Datos hematológicos y estudio molecular de las familias I y II
| Parámetros | Familia I | Familia II | ||||||
| Abuelo | Padre | Madre | Propositus | aI1 (padre) | I2 (madre) | aII1 (propositus) | II2 (hermana) | |
| Sexo/edad (años) | M/65 | M/38 | F/32 | M/6 | M/45 | F/40 | F/16 | F/8 |
| Genotipo | --/αα | --/αα | αα/αα | --/αα | -α3.7/αα | --/αα | --/-α3.7 | αα/αα |
| Hematíes, 1012/L | 6,5 | 5,5 | 4,7 | 5,5 | 5,1 | 5 | 5 | 4,6 |
| Hto (%) | 43,2 | 38,4 | 41,7 | 36,6 | 43,8 | 36 | 31,2 | 42 |
| Hb (g/dl) | 13,2 | 12,4 | 14 | 11,6 | 15,2 | 11,7 | 9,4 | 14,6 |
| VCM (fl) | 66,3 | 69,7 | 88,1 | 67,4 | 86,3 | 71,9 | 61 | 91 |
| HCM (pg) | 20,2 | 22,5 | 29,6 | 21,2 | 29,9 | 23,4 | 18,7 | 31,7 |
| RDW, % | 15,6 | 14,3 | 13,2 | 15,3 | 12,4 | 13,1 | 18,1 | 12,5 |
| Reticulocitos, % | 0,8 | 1 | 1 | 1 | 0,6 | 0,8 | 1,9 | 0,6 |
| Cuerpos de inclusión | + | + | - | + | - | + | +++ | - |
| HbA2, % | 3,3 | 1,3 | 1 | 0,8 | 1,9 | 1,8 | 0,8 | 2,1 |
| HF, % | 0,4 | 1,0 | 1,9 | 1,8 | 0,8 | 1 | 1,7 | 0,4 |
| Hb H, % | 0 | 0 | 0 | 0 | 0 | 0 | 8 | 0 |
| Hipocromía | ++ | ++ | Normal | ++ | Normal | ++ | +++ | Normal |
| 3’HVR (Puv II) | 3 | 4,4 | 3,4; 1,9 | 3,4 | 5,5; 4 | 5 | 4 | 5,5; 5 |
| 5’HVR (Rsa I) | 6; 2 | 2,2; 2 | 3,8; 3 | 3,8; 2 | 9; 2 | 3 | 9 | 3; 2 |
Hb: hemoglobina; HCM: hemoglobina corpuscular media; HF: hemoglobina fetal; Hto: hematocrito; VCM: volumen corpuscular medio.

En la segunda familia, los datos de PCR y FISH mostraron que en el extremo 5’ se produjo una deleción subtelomérica entre los plásmidos 33803 y 37123, dejando un punto de corte de diferencia de aproximadamente 3,3kb.
En el extremo 3’ centromérico, el punto de corte de la deleción se mostró entre los plásmidos p 177265 y 178649, mostrando una diferencia de exactitud en la deleción de aproximadamente 1,4 kb. En este segundo caso la deleción α (--GP) es de 145 Kb (fig. 1).
DiscusiónEl gen de globina alfa está situado en una zona muy activa del cromosoma 16p, cerca del telómero (-150kb). Se ha secuenciado un largo segmento del cromosoma 16 de alrededor de 500 Kb, desde la región telomérica repetitiva hasta el centrómero. Ello ha servido para analizar la replicación, la metilación de los genes alfa, así como el desarrollo estructural de la cromatina nuclear. Además, han sido estudiadas diferentes mutaciones de α talasemia, que han permitido comprender las conexiones que existen entre la estructura del gen y su función.
Estudios epidemiológicos realizados en nuestro país han demostrado que la frecuencia alélica de la α talasemia es del 4,79%, más frecuente que lo que se observa en la β talasemia (0,1-2%)7. La deleción más frecuente es la pérdida de 3,7kb de ADN, dejando un solo gen funcional (-α3.7/αα). Mucho menos frecuentes son las mutaciones de 4,2kb y las deleciones de ambos genes α en el mismo cromosoma (α° talasemias heterocigotas).
En este trabajo hemos caracterizado los puntos de ruptura de los extremos 5’ y 3’ de dos familias españolas con dos nuevas mutaciones de α° talasemia, no descritas hasta la actualidad en la literatura mundial.
Los puntos de ruptura de las deleciones se caracterizan por una combinación de Southern blot, análisis de PCR y FISH. En ambos casos la mutación deleciona ambos genes α y la región reguladora HS-40. En la primera familia, la mutación --ED ocupa aproximadamente 80kb con el punto de ruptura 5’ en la coordinada +100 (± 3kb), mientras que en el extremo 3’HVR se encuentra situado en la coordenada +178±0,75kb.
La segunda mutación, - -GP, ocupa una región más extensa, de 145kb, localizándose el extremo 5’ entre las coordenadas 34 y 37, y la región centromérica en la coordenada +178±1,4. Es decir, ambas mutaciones, - -ED y - -GP, se sitúan próximas en la región centromérica. El propositus II1 y su madre I2 tienen una inteligencia normal, no presentan dismorfias y desde el punto de vista fenotípico son completamente normales. Esta mutación es subtelomérica, respetando por lo tanto el telómero, a diferencia de lo que sucede con otras dos mutaciones de origen español: - -BR y - -CMO. En ambas se afecta el telómero y en ambas existe truncación con reposición del telómero con secuencias teloméricas repetitivas8,9.
También hemos estudiado estas mutaciones con MLPA, pero con este método no hemos sido capaces de delimitar específicamente los extremos 3’ y 5’. Sin embargo, la combinación de Southern, PCR y FISH, de una mayor complejidad, puede delimitar con mayor precisión los puntos de corte de las deleciones. La simplicidad de la técnica de MLPA en relación con las otras técnicas puede ser utilizada para localizar las lesiones en una primera etapa1 y finalmente delimitar los puntos de ruptura por FISH y PCR6,10.
En resumen, en este trabajo presentamos dos nuevas mutaciones de α° talasemia, con una paciente con enfermedad de la Hb H. En ambas se suprime la región 3’HVR y la región regulador H40, y en la segunda también la región 5’HVR.
En España la inmigración supone un 10% de la población y en Madrid unos 800.000 habitantes son extranjeros. Por esta razón es de interés el estudio de estas nuevas deleciones, dado que la asociación de estas nuevas mutaciones, con casos de hemoglobinopatías α y α talasemias, frecuentes en la población inmigrante, sobre todo la mutación –SEA en la población asiática11,12, pueden modificar el genotipo de futuras generaciones y ser de mucha utilidad para establecer el consejo genético13.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Al Prof. D.R. Higgs, del MRC Molecular Institute, John Radcliffe Hospital, Universidad de Oxford, Reino Unido, por su ayuda y por la donación de los plásmidos.
