




El retraso mental, caracterizado por un funcionamiento intelectual significativamente inferior a la media y referido a limitaciones sustanciales en el desenvolvimiento diario, se presenta en la población general con una frecuencia del 1 al 3%1,2. A pesar de las investigaciones clínicas y de laboratorio que se realizan, sólo se identifican factores etiológicos específicos3 en aproximadamente la mitad de los pacientes.
Desde el año 1995 está bien definido que alteraciones en las regiones subteloméricas cromosómicas explicarían la etiopatogenia del trastorno del desarrollo/retraso mental en algunos casos hasta ahora sin causa identificable4. Fenotipos específicos asociados con determinadas alteraciones subteloméricas comienzan a emerger y ser clínicamente reconocibles5. Algunos ejemplos de deleciones subteloméricas ampliamente descritas son la deleción 1pter, la deleción 1qter o el síndrome de deleción 5q35.1. Sin embargo, otros síndromes que afectan a estas regiones cromosómicas se describen con menor frecuencia en la bibliografía, como es el caso del síndrome de deleción 9q34. Esta alteración se ha descrito recientemente en varios pacientes, lo que ha dado lugar a la definición de un nuevo síndrome emergente: el síndrome del 9qter.
Presentamos 2 pacientes con retraso mental, dismorfia facial y anomalías congénitas, en los que se detectó una deleción subtelomérica y submicroscópica en el brazo largo del cromosoma 9.
Pacientes y método
Caso 1
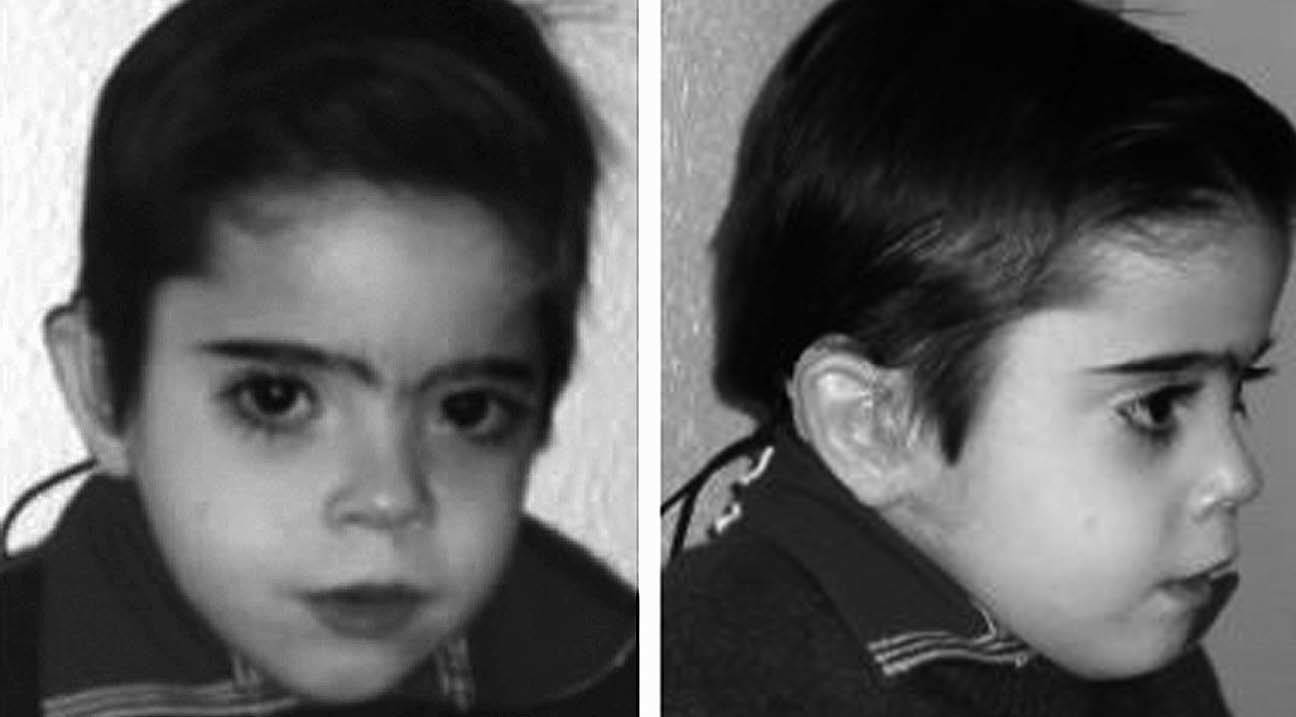
El primer paciente es el quinto hijo de una pareja no consanguínea y sin antecedentes familiares de interés. Durante el segundo mes de embarazo, la madre fue intervenida de un carcinoma de tiroides y a partir de entonces recibió tratamiento hormonal sustitutivo. Se realizó una amniocentesis en un laboratorio externo, cuya indicación fue la edad materna avanzada, con resultado 46,XY (cariotipo normal de varón). La gestación transcurrió sin ninguna otra incidencia y finalizó a las 37 semanas tras un parto natural. El paciente presentó los siguientes parámetros neonatales: 43 cm de estatura, 1.975 g de peso y 29,5 cm de perímetro craneal. El test de Apgar fue de 9/10. El paciente nació con manifestaciones derivadas de los efectos del tratamiento con hormonas tiroideas que había recibido la madre, como intranquilidad y nerviosismo, entró en parada cardiorrespiratoria y a partir de ese momento presentó hipotonía muscular generalizada. Presentaba facies plana y rasgos dismórficos como boca en carpa, hipertelorismo, fisuras palpebrales hacia abajo, sinofridia, orejas de implantación baja y prognatismo, así como macroglosia con asimetría de la lengua, que le ocasionó problemas de alimentación durante el período neonatal (fig. 1). Como anomalías congénitas cabe destacar la persistencia del conducto peritoneo vaginal, intervenido durante la infancia. El desarrollo somático fue normal.
Fig. 1. El caso 1 presenta facies plana y rasgos dismórficos como boca en carpa, hipertelorismo, fisuras palpebrales hacia abajo, sinofridia, orejas de implantación baja y prognatismo. (Imágenes reproducidas con permiso).
Respecto a los rasgos neurológicos, presenta un retraso mental leve-moderado y desarrollo psicomotor alterado (sostén cefálico a los 6 meses; sedestación en torno a los 12-15 meses; inicio de la marcha con ayuda a los 18 meses; en la actualidad, con 4 años, empieza a pronunciar alguna palabra suelta). Además, desde el punto de vista neurológico, presenta hipoacusia neurosensorial y bilateral, que le obliga a llevar audífonos, y persiste la hipotonía neonatal. Presenta estereotipias, realizando movimientos de torsión con las manos. Las exploraciones complementarias realizadas, como la resonancia magnética, no muestran alteraciones significativas. En cuanto al comportamiento, es un niño sociable y feliz, que sonríe con mucha facilidad.
Caso 2
La segunda paciente es la cuarta hija de una pareja no consanguínea y con el antecedente familiar de un tío paterno diagnosticado de parálisis infantil al nacimiento. No se registró ninguna incidencia durante el embarazo y, dado que no había ninguna indicación, no se realizó diagnóstico prenatal citogenético. La gestación finalizó a las 31 semanas con una cesárea urgente por sufrimiento fetal y presentación transversa. La paciente presentó los siguientes parámetros neonatales: 42 cm de estatura, 1.800 g de peso y 28 cm de perímetro craneal. El test de Apgar fue de 7/9. Al nacer ingresó en neonatos por facies tosca, fenotipo anómalo y pie equino varo izquierdo reductible. A los pocos días se auscultó un soplo sistólico que parecía de características funcionales. Durante su ingreso en neonatos presentó anemia del prematuro, que precisó transfusiones de concentrados de hematíes, y traquipnea transitoria del recién nacido. Durante los primeros meses de vida presentó neumo-nías de repetición y episodios broncoobstructivos que precisaron numerosos ingresos hospitalarios. La paciente presentaba microcefalia, facies plana y rasgos dismórficos como hipertelorismo, fisuras palpebrales hacia abajo, sinofridia, orejas de implantación baja, nariz pequeña con la raíz nasal ancha y narinas antevertidas, boca en carpa, tendencia a la protusión lingual y diástasis dentaria. Muestra un babeo continuo. El desarrollo somático fue normal, y tan sólo mostró manos toscas y cortas.
Respecto a los rasgos neurológicos, presenta retraso mental grave y desarrollo psicomotor alterado (sostén cefálico con sedestación a los 10 meses; inicio de la marcha libre hacia los 5 años; en la actualidad, con 11 años, no habla, tan sólo balbucea). Además, desde el punto de vista neurológico presenta hipoacusia neurosensorial bilateral, que le obliga a llevar audífonos, e hipotonía muscular. Presenta movimientos de balanceo y estereotipias en manos y pies. Desde siempre manifiesta alteraciones del sueño, despertares bruscos y sueño interrumpido, que ha precisado en alguna ocasión tratamiento farmacológico. Las exploraciones complementarias efectuadas resonancia magnética cerebral, tomografía computarizada, estudio bioquímico de enfermedades metabólicas congénitas (aminoácidos, ácido láctico, ácido pirúvico, etc.) no muestran hallazgos patológicos. Por lo que se refiere al comportamiento, es una niña indiferente al medio que la rodea y con tendencia a autolesionarse.
Método
El estudio cromosómico se realizó con técnicas de citogenética convencional y bandas GTG con una resolución de 450-500 bandas. Se utilizó la técnica de MLPA (multiplex ligation-dependent probe amplification) con sondas específicas para las regiones subteloméricas de todos los cromosomas (SALSA P036B y P070, MRC-Holland, Amsterdam, Países Bajos). Esta técnica permite analizar desequilibrios de la dosis génica para todos los telómeros de los cromosomas, a excepción de los brazos cortos de los acrocéntricos (13, 14, 15, 21 y 22). La técnica se realizó siguiendo las recomendaciones de los fabricantes con pequeñas modificaciones descritas previamente6.
Para la técnica de FISH (hibridación in situ fluorescente) se emplearon sondas de las regiones subtelomérica (ToTelVysion TM Multi-Color, VYSIS, Downers Grove, IL, EE.UU.). El estudio con esta técnica se realizó siguiendo el protocolo recomendado, salvo algunas modificaciones7. Después de la hibridación se evaluaron como mínimo 10 metafases utilizando el microscopio Nikon Eclipse E6000 (Tokio, Japón) y el programa Quips R Imaging (VYSIS, Downers Grove, IL, EE.UU.).
Para delimitar la región delecionada se realizó un análisis de segregación con 3 marcadores microsatélites desarrollados por nuestro grupo, que se localizan en las posiciones 136.550, 137.200 y 137.350 kb (oligonucleótidos a disposición de quien lo solicite), mediante amplificación por reacción en cadena de la polimerasa y electroforesis en geles de poliacrilamida (12%) no desnaturalizante. Estos marcadores microsatélites se localizan en los clones LL09NC01-272H12 y RP11-413M3. Las secuencias se obtuvieron del NCBI (http://www.ncbi.nlm.nih.gov/mapview/ map_search.cgi).
Resultados
En el caso 1 se realizó amniocentesis materna por edad avanzada, con resultado 46,XY (cariotipo normal de varón). Dadas las manifestaciones clínicas que presentaba el paciente, se repitió el cariotipo a los 2 años de edad y se obtuvo el mismo resultado que en la amniocentesis. En el caso 2, a los 10 meses de vida fue remitida para la realización de un cariotipo debido al retraso psicomotor evidente y el fenotipo anómalo. El resultado fue 46,XX (cariotipo normal de mujer).
En ambos casos el estudio mediante la técnica de MLPA permitió identificar una disminución de dosis en la región terminal de los brazos largos del cromosoma 9, concretamente en 9q34. Este resultado se confirmó en los 2 pacientes mediante la técnica de FISH con sondas subteloméricas específicas del cromosoma 9. Los resultados mostraron una deleción subtelomérica en los brazos largos de dicho cromosoma.
Estos mismos estudios se realizaron a los padres de ambos pacientes, con resultados normales, lo cual indica que la alteración detectada en los pacientes surgió de novo.
Con el fin de determinar el tamaño de la región delecionada y conocer su origen, se realizó el análisis de segregación por medio de marcadores microsatélites. En el caso 1 se observó pérdida del alelo paterno en 137.350 y segregación normal en 136.550. El marcador 137.200 no fue informativo. Se dedujo que la región delecionada era menor de 1,5 Mb. En el caso 2 se observó pérdida del alelo paterno en el marcador 137.350 y segregación normal en 137.200. Se dedujo que la región delecionada era menor de 0,8 Mb.
Discusión
La deleción submicroscópica del(9)(q34) es un síndrome de microdeleción poco frecuente, con unos 33 casos descritos en la literatura científica8-16. Las deleciones en la región 9q34 visibles citogenéticamente son muy muy raras10,17,18, posiblemente porque grandes pérdidas de material genético en esta región son letales en etapas tempanas de la embrioge-nia19 o producen el fallecimiento del recién nacido en torno a los 3-5 meses de edad17,18 por fallo cardiorrespiratorio.
El síndrome de deleción 9q34 parece ser una entidad clínicamente reconocible. Por lo general, los pacientes presentan rasgos dismórficos comunes como facies plana con hipertelorismo, sinofridia, nariz pequeña con narinas antevertidas, boca en carpa con protusión lingual, junto con hipotonía, retraso mental y anomalías congénitas, sobre todo defectos cardíacos conotruncales. Con menor frecuencia se encuentran otras manifestaciones clínicas, como obesidad, pérdida de audición, anomalías urogenitales, trastornos del sueño y problemas de comportamiento.
La deleción 9qter puede producirse por una translocación cromosómica críptica, que puede encontrarse equilibrada en uno de los progenitores o aparecer de novo, o bien por reordenamientos intracromosómicos que darían lugar a deleciones puras. La mayoría de los casos descritos en la literatura médica suelen corresponder a deleciones puras (14:22), al igual que los 2 casos presentados. En los casos originados por una translocación subtelomérica críptica en los que hay pérdidas y ganancias de material cromosómico parece que predominan las manifestaciones clínicas debidas a la deleción frente a las que se producirían por la trisomía.
La región delecionada puede ser variable en cuanto a tamaño, generalmente de 1 a 2,3 Mb, con unos 20 genes descritos en esta zona. La deleción más pequeña asociada a este síndrome8, con un tamaño en torno a 700 kb, incluía varios genes candidatos: ZMYND19, ARRDC1, C9ORF37, EHMT1 y CACNA1B. Sin embargo, en un estudio publicado recientemente16, al mismo tiempo que se identifica la deleción 9qter más pequeña (380 kb), se establece que la haploinsuficiencia del gen EHMT1 (euchromatin histone methyltransferasa-1) es la causa del síndrome de deleción 9qter, por la presencia de 2 mutaciones deletéreas de novo en pacientes con el fenotipo característico. EHMT1 es una metiltransferasa de histonas e interviene en la remodelación de la cromatina que participa en la neurogenia y proliferación neuronal durante el desarrollo embrionario, y se expresa en tejido cerebral y músculo cardíaco en el adulto8,12. Significativamente, un número creciente de genes implicados en el retraso mental sindrómico participan igualmente en procesos de remodelación cromatínica, como los genes implicados en los síndromes de Rett, de Coffin-Lowry, de ATR-X (síndrome de alfatalasemia y retraso mental ligado al cromosoma X), etc.
La gravedad del fenotipo de este síndrome se relacionó con la extensión de la deleción8,13,17 y, de forma más discutida, con la implicación de los posibles genes candidatos en dicha región8. No obstante, la relación genotipo-fenotipo no parece mantenerse en el caso de nuestros pacientes, ya que, al presentar el caso 2 una deleción menor que el caso 1, parece que su afectación clínica y su pronóstico son peores. Un ejemplo de esta mayor gravedad clínica se refleja en la susceptibilidad a episodios broncoobstructivos e infecciones, como neumonías de repetición, típicos de este síndrome, que presenta únicamente el caso 2. Estos problemas respiratorios pueden deberse a trastornos aspirativos broncopulmonares, a la hipotonía o a una deficiencia inmunitaria.
De los aproximadamente 35 casos descritos con síndrome de deleción 9q34, tan sólo 613,17,20 presentan pérdida de audición o sordera, como ocurre en los 2 casos que presentamos. En 9q34.3 se ha descrito un locus (DFNB33) relacionado con sordera neurosensorial, de carácter autosómico recesivo, en una familia jordana en la que varios miembros presentan sordera y en la que hay antecedentes de consanguinidad. Su posible relación con este síndrome parece discutible.
Los pacientes con este síndrome presentan varios grados de afectación cardíaca congénita. Los defectos más comunes suelen ser malformaciones cardíacas conotruncales, como defecto septal ventricular, coartación de aorta, estenosis o displasia valvular y tetralogía de Fallot. El hecho de que nuestros pacientes no manifiesten anomalías cardíacas puede indicar un mejor pronóstico, ya que la causa de muerte en la mayoría de pacientes con este síndrome es el fallo cardiorrespiratorio.
En cuanto al origen parental de la deleción, en la mayoría de los casos publicados la deleción afecta al cromosoma de origen paterno (5:6). Incluidos los 2 casos aquí descritos, dicha proporción (7:8) resulta significativa (*2 = 4,5; p = 0,03), por lo que podemos concluir que la deleción 9qter pura tiende a surgir en la línea germinal masculina durante la espermatogenia. Así, la deleción 9qter se suma a otros síndromes con alteración genómica de aparición preferente en la línea germinal masculina, como son la enfermedad de Pelizaeus-Merzbacher21 o la neuropatía Charcot-Marie-Tooth tipo 122. Es importante identificar posibles factores de riesgo predisponentes a reordenamientos subteloméricos con el fin de protocolizar adecuadamente el cribado prenatal. Asimismo, es importante identificar el origen, hereditario o no, de la deleción para garantizar un adecuado consejo genético.
