




Se describe una nueva mutación en el gen δ-globina (delta-talasemia), responsable de una disminución de los valores de hemoglobina (Hb) A2 y asociada a Hb Watts, variante de Hb debido a una deleción de trinucleótidos.
Pacientes y métodoEl análisis de Hb se llevó a cabo mediante high performance liquid chromatography (HPLC, «cromatografía líquida de alta resolución») de intercambio iónico y electroforesis capilar de zona. Se utilizaron técnicas de reacción en cadena de la polimerasa y secuenciación automática para identificar las mutaciones en los genes δ- y α-globina.
ResultadosLa Hb anómala se observó en la electroforesis capilar de zona en Z6 y por HPLC de intercambio iónico apareció un pico más lento que la HbA en un tiempo de retención de 4,19min. Esta variante de la Hb se llama Hb Watts [α2 74(EF3)Asp->0 o α2 75(EF4)Asp->0; HBA2:c.226_228delGAC]. El bajo porcentaje de HbA2 se debe a una inserción de 27nt entre los nucleótidos 83 y 84 de IVS-I del gen de δ-globina.
ConclusionesCuando se analiza un cromatograma se debe tener en cuenta la posibilidad de una delta-talasemia o una variante de HbA2, aparte de una alfa-talasemia, beta-talasemia y hemoglobinopatías estructurales. A tal fin, cada uno de los picos y sus porcentajes deben ser considerados para una correcta interpretación y evitar diagnósticos erróneos tanto como sea posible.
We describe a novel delta-thalassemia mutation causing decreased hemoglobin (Hb) A2 levels associated with Hb Watts, variant Hb resulting from a trinucleotide deletion in Spain.
Patients and methodHb variant analysis was performed by cation-exchange high performance liquid chromatography (HPLC) and capillary zone electrophoresis. Polymerase chain reaction and DNA sequence analyses were used to identify mutations in the δ- and α-globin genes.
ResultsAbnormal Hb was observed on capillary zone electrophoresis in Z6 and by cation-exchange HPLC a slower peak than HbA was observed at an retention time of 4.19min. This variant Hb is called Hb Watts [α2 74(EF3)Asp->0 or α2 75(EF4)Asp->0; HBA2:c.226_228delGAC]. The decreased HbA2 percentage owes to an insertion of 27nt between nt 83 and 84 of IVS-I of the δ-globin gene.
ConclusionsWhen analyzing a chromatogram, the possibility of the existence of delta-thalassemia or an HbA2 variant should be considered, apart from alfa-, beta-thalassemia and structural haemoglobinopathies. To this end, each of the peaks and their percentages should be considered to allow for correct interpretation and to avoid misdiagnosis as much as possible.
Las principales hemoglobinas (Hb) en los humanos adultos son la HbA y HbA2. Ambas están constituidas por cadenas de globina α asociadas a cadenas β (α2β2) o cadenas δ (α2δ2), respectivamente. Los genes que controlan la síntesis de las cadenas δ y β se encuentran localizados en el brazo corto del cromosoma 11, participando en el cluster β de globina, mientras que los genes para las cadenas α se encuentran en el cromosoma 161. La HbA2 no tiene funciones bioquímicas específicas y, en condiciones normales, su fracción puede ir de 2,0-2,22 a 3,2-3,4%3,4 del total de la Hb. Defectos en el gen δ-globina pueden modificar la expresión de la fracción HbA2, disminuyendo o aumentando el porcentaje de la misma. Valores inferiores al 2,0% son generalmente debidos a defectos genéticos que causan reducción de la síntesis de la cadena δ (delta-talasemia), síntesis de variantes de HbA2, o bien cundo se presenta una alfa+-talasemia homocigota, alfa0-talasemia heterocigota, enfermedad de la Hb H y algunos casos de delta-beta0-talasemia. Por el contrario, los valores aumentados de HbA2 están relacionados, entre otros, con la beta-talasemia heterocigota, rasgo de células falciformes y Hb inestables5.
Describimos una nueva mutación en el gen δ-globina (delta-talasemia), responsable de una disminución de los valores de HbA2 y asociada a Hb Watts, variante de Hb debida a una deleción de trinucleótidos.
Paciente y métodoLa propositus es una mujer asintomática de 58 años de edad, natural de Jerez de la Frontera, que fue sometida a un control de diabetes debido a su obesidad. El análisis del hemolizado para la Hb glucosilada utilizando una técnica de high performance liquid chromatography (HPLC, «cromatografía líquida de alta resolución») (Variant™ II; Bio-Rad Laboratories, Hercules, CA, EE. UU.) reveló un pico inusual en un retention time (RT, «tiempo de retención») de 1,14min.
Una muestra de sangre fue remitida al Centro de Referencia de Talasemia y Hemoglobinopatías (Hospital Clínico San Carlos) para su caracterización molecular. Los datos hematológicos fueron obtenidos en un contador de células automatizado (Coulter® LH750 Analyser, Beckman Coulter, Brea, CA, EE. UU.). Los valores de HbA2 y Hb fetal (Hb F) fueron medidos por HPLC-cation-exchange (HPLC-CE, «HPLC de intercambio iónico») (Variant™). Las Hb fueron estudiadas por electroforesis capilar de zona (ECdZ) (Sebia Capillarys® Flex; Sebia, Norcross, GA, EE. UU.) y HPLC-CE utilizando el programa corto para la beta-talasemia de Bio-Rad (Bio-Rad, Hercules, CA, EE. UU.), que separa las variantes de Hb usando un gradiente salino. Las cadenas de globina fueron analizadas por HPLC de fase reversa6.
Tras el aislamiento de ADN genómico con un método automático (BioRobot® EZ1; Qiagen GmbH, Hilden, Alemania), el ADN fue cuantificado con un NanoDrop® 1000 (Thermo Scientific, Wilmington, DE, EE. UU.).
Las mutaciones más frecuentes para la alfa-talasemia fueron estudiadas por reacción en cadena de polimerasa múltiple Alpha-Globin StripAssay® (ViennaLab Diagnostics GmbH, Viena, Austria).
La secuenciación de los genes α se llevó a cabo siguiendo el protocolo publicado por De la Fuente-Gonzalo et al.7. El gen de la δ-globina también fue analizado directamente por secuenciación; en este caso fue necesario amplificar 2 segmentos, como ha sido descrito con anterioridad8.
Todos nuestros índices hematológicos y hallazgos clínicos fueron recogidos previo consentimiento informado de la propositus y aprobación por el Comité de Ética del Hospital Clínico San Carlos.
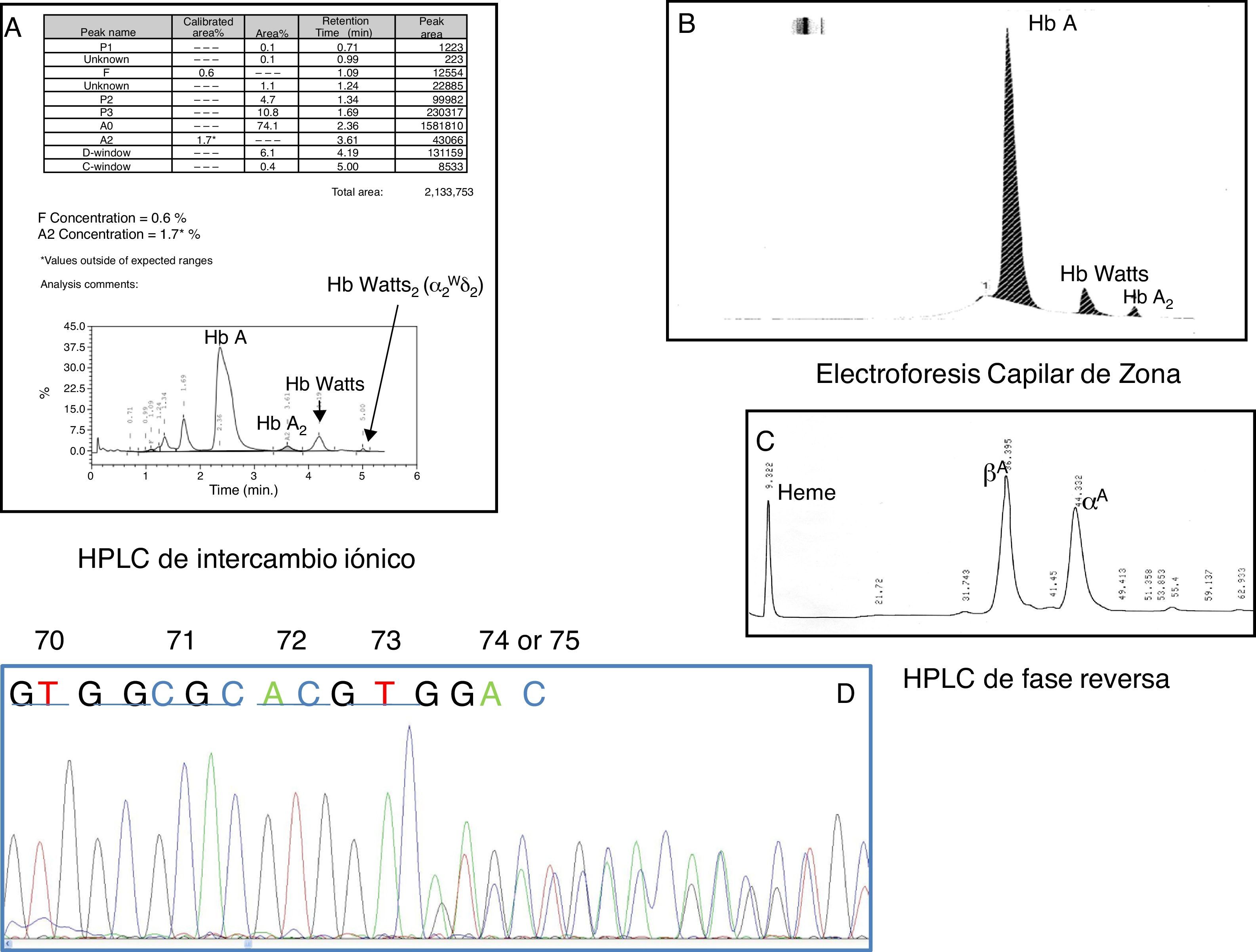
ResultadosLos datos hematológicos (Hb 15,4g/dl, hematocrito 0,50l/l, hematíes 5,59×1012/l, volumen corpuscular medio 90,3fl, Hb corpuscular media 27,5pg e índice de amplitud eritrocitaria 12,6%), el frotis de sangre periférica y el metabolismo férrico fueron normales. Durante la cuantificación de la HbA2 (1,7%) y la Hb F (0,6%) en la HPLC-CE apareció un pico más lento que en la HbA en RT de 4,19min (D-window) y que se correspondió con el 6,1% del total de la Hb, y un segundo pico en un RT de 5min de solo el 0,4% de la Hb total (fig. 1A). Mediante ECdZ una Hb anómala fue observada en Z6, cuya concentración fue del 6,9% (fig. 1B). El estudio de las cadenas de globina por HPLC de fase reversa no reveló picos anómalos (β y α). Dado el porcentaje de Hb anómala, esta podría ser una variante de cadena α. Para confirmar esta idea, la secuenciación del ADN del gen α2 de globina mostró 3 nucleótidos (GAC) suprimidos entre los codones 74 y 75, correspondiente al ácido aspártico; esta variante de Hb se denomina Hb Watts [alfa2 74(EF3)Asp->0 o alfa2 75(EF4)Asp->0; HBA2:c.226_228delGAC] (fig. 1C).
![A: Cromatografía líquida de alta resolución de intercambio iónico. B: Electroforesis capilar de zona. C: Cromatografía líquida de alta resolución de fase reversa. D: Secuenciación directa automatizada del exón 2 del gen α2 de globina mostrando la deleción GAC entre los codones 74 y 75 correspondiente a la Hb Watts [α2 74 (EF3) Asp-> 0 o α2 75 (EF4) Asp-> 0; HBA2:c.226_228delGAC]. HPLC: high performance liquid chromatography («cromatografía líquida de alta resolución»).](https://static.elsevier.es/multimedia/00257753/0000014400000007/v2_201503121238/S0025775314008136/v2_201503121238/es/main.assets/gr1.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNeMnx59J5MZq19QkIYapO3CmCEVynax+YwwiqXCUMrK9w1j33FY+c+LTQapYvzn/Jjm9yJ54t/RXiLkqEti/cUuOyXKWdl1y/tOzfCRYtsR1ulilccgML8fHEAnNCVh8ZXZZGDR5dUhWaBLlJC5XeUy+Ak6bPzOxB+QQzrGEbZudYrf/0xOnkHiFNHvNjXaVUbDjWI8DJZhakMwxo7edVdhNQDqYgmyN0IuSIJDAXG2KuV3QRIUSBel3PRyvnsa2/0= "A: Cromatografía líquida de alta resolución de intercambio iónico. B: Electroforesis capilar de zona. C: Cromatografía líquida de alta resolución de fase reversa. D: Secuenciación directa automatizada del exón 2 del gen α2 de globina mostrando la deleción GAC entre los codones 74 y 75 correspondiente a la Hb Watts [α2 74 (EF3) Asp-> 0 o α2 75 (EF4) Asp-> 0; HBA2:c.226_228delGAC]. HPLC: high performance liquid chromatography («cromatografía líquida de alta resolución»).")
A: Cromatografía líquida de alta resolución de intercambio iónico. B: Electroforesis capilar de zona. C: Cromatografía líquida de alta resolución de fase reversa. D: Secuenciación directa automatizada del exón 2 del gen α2 de globina mostrando la deleción GAC entre los codones 74 y 75 correspondiente a la Hb Watts [α2 74 (EF3) Asp-> 0 o α2 75 (EF4) Asp-> 0; HBA2:c.226_228delGAC].
HPLC: high performance liquid chromatography («cromatografía líquida de alta resolución»).
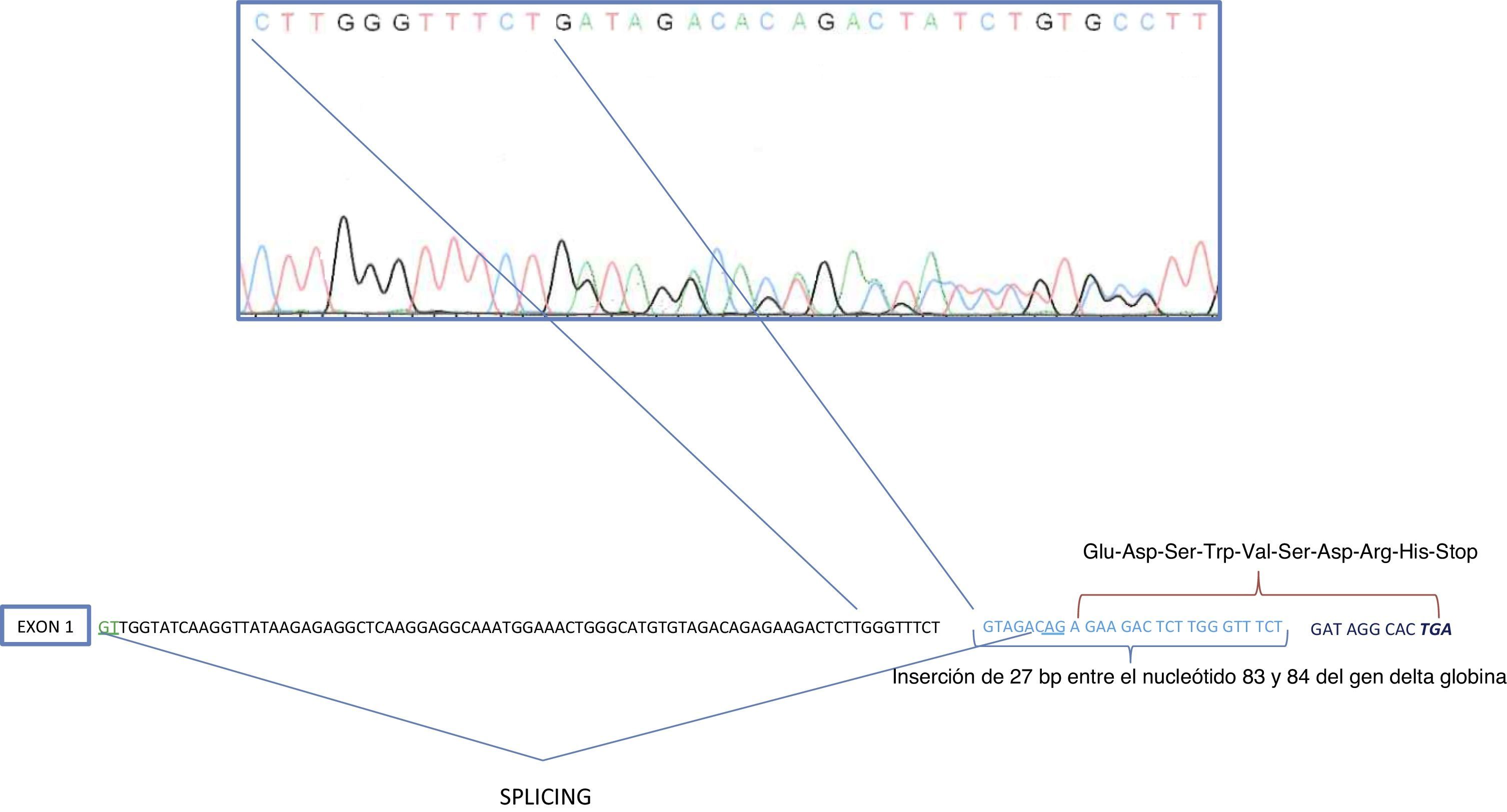
Debido a la disminución del porcentaje de HbA2 y al pequeño pico por HPLC-CE a un RT de 5min, se llevó a cabo la secuenciación del gen δ-globina, que mostró una inserción de 27 pares de bases (pb) entre los nucleótidos 83 y 84 de IVS-I (GTAGACAGAGAAGACTCTTGGGTTTCT). Esta anomalía de 27pb coincide con la secuencia que hay entre el IVS-I-nt-56 y el IVS-I-nt-83 del gen δ-globina (fig. 2).
.")
Arriba: Secuenciación directa automatizada del gen δ-globina. Frameshift debido a la inserción de 27 pares de bases entre los nucleótidos 83 y 84 de IVS-I. Parte inferior: Simulación de sitio alternativo para el splicing originado por el frameshift y la secuencia de los nuevos residuos de aminoácidos (Glu-Asp-Ser-Trp-Val-Ser-Asp-Gln).
Tanto la alfa-talasemia no deleción como la deleción fueron descartadas.
DiscusiónLas alteraciones en la cadena δ de globina no tienen significación clínica y el único parámetro hematológico que parecen alterar son los valores de HbA2. En el caso de heredarse junto con una beta-talasemia heterocigota, donde los valores de HbA2 suelen estar aumentados, es la causa más común de normalización de la HbA2. En este trabajo se presenta una nueva delta0-talasemia asociada a una hemoglobinopatía estructural de cadena α (Hb Watts), en donde se reitera el descenso de los valores de HbA2.
La Hb Watts fue descrita por primera vez en 1997 en una mujer de 37 años de origen mexicano, pero nacida en Watts (Los Angeles Country, CA, EE. UU.)9; desde entonces no ha vuelto a ser publicada. La Hb Watts es una variante de cadena α globina y una de las pocas debida a la deleción de pb. Aunque la zona donde se localizan los residuos 74 y 75 de la cadena α2 globina no es una región crítica de la molécula de Hb, la deleción de cualquiera de estos residuos parece originar una ligera inestabilidad en la molécula, sin manifestaciones clínicas ni alteraciones hematológicas. Fue detectada durante un control de diabetes, pero no afecta al valor de la Hb glucosilada, por lo que el valor diagnóstico de la prueba no se ve alterado.
Cuando existe una variante estructural de Hb, ya se trate de cadena α1 como de α2, suelen presentar una HbA2 algo disminuida (≈2,2%), porque en la mayoría de ellas se forma su correspondiente variante de HbA210. Sin embargo, en el caso de esta Hb Watts, los valores de HbA2 eran más bajos de lo habitual (1,7%), y dado que también se observó un pico anómalo por HPLC-CE a un RT de 5min, probablemente podría existir alguna anomalía en las cadenas δ de globina.
La secuenciación del gen δ-globina reveló una inserción de 27pb entre los nucleótidos 83 y 84 del IVS-I, que en realidad se correspondía con una duplicación de la secuencia comprendida entre los nucleótidos 56 y 83 del mismo intrón. Esta duplicación proporciona una nueva secuencia consenso «AG» para el splicing del primer intrón, adelantándose aproximadamente unos 30bp. A la vez se produciría un frameshift, de modo que 28bp hacia delante del splicing alternativo se originaría un codón de stop (fig. 2). Esta nueva lectura codificaría una nueva proteína de 40 aminoácidos, los 31 primeros coinciden con los de la cadena δ globina y el resto vendrían establecidos por la nueva secuencia (Glu-Asp-Ser-Trp-Val-Ser-Asp-Gln). La proteína resultante sería inestable y, por lo tanto, se trataría de una delta0-talasemia, lo que originaría la disminución de HbA2.
Se han descrito un total de 110 mutaciones que afectan al gen δ-globina, 67 originan variantes estructurales de HbA2 y el resto corresponden a delta-talasemia11. La mayoría de los defectos génicos responsables de las delta-talasemias que alteran la expresión del gen δ-globina se deben a la sustitución de un único nucleótido (mutación puntual), y en menor medida a grandes deleciones y entrecruzamiento de genes. Es interesante que el 65% de las lesiones que causan alguna variante estructural de cadena δ tienen su equivalente en el gen β y el 35% de la delta-talasemia son las mismas mutaciones que causan la beta-talasemia. Se cree que estas sustituciones de nucleótidos idénticos han surgido ya sea como mutaciones independientes, o como resultado de episodios de conversión de genes1. La mayor parte de las beta-talasemias se ha detectado debido a su asociación con una beta-talasemia en trans, como la deleción Corfú (−7,2kb) asociada con la mutación sin sentido CD39 (C>T)12, o en cis, como por ejemplo la pérdida de una A en el CD59 del gen δ y la Hb Knossos [β27 (B9) Ala> Ser; HBB:c.82G> T]13. Esta nueva mutación es la primera que se produce mediante la inserción de una secuencia de 27pb.
Una vez identificada la causa del descenso de la HbA2 quedaba interpretar el pico que aparecía en la C-window por HPLC-CE. Quedó descartado que se tratara de la HbA’2 [δ16(A13) Gly>Arg; HBD:c.49G>C], ya que esta por HPLC eluye en la S-window14–16, de modo que podría corresponder a la HbA2 formada por la variante de cadena αWatts. Dicha Hb no fue detectada por electroforesis capilar, y tampoco en la publicación de 1997 sobre la Hb Watts se hablaba sobre ninguna otra HbA2. Ello podría ser debido a que su bajo porcentaje hace que sea difícil de discernir en la electroforesis de Hb y, aunque es fácilmente detectable por HPLC-CE, todavía parece que hay una falta de educación sobre las variantes de HbA2.
Se corrobora que la delta-talasemia no tiene significación clínica, ya que el ser heredada junto con una variante de Hb de cadena α no se observaron parámetros hematológicos alterados, tan solo el descenso en los valores de HbA2. En estos casos hay que tener en cuenta que se podría formar la variante de HbA2 correspondiente, aunque el porcentaje sería tan pequeño que por algunas de las técnicas electroforéticas no se detectaría16. El valor real de la HbA2 lo constituiría la suma de ambas concentraciones.
En los casos de coexistencia de delta-talasemia en cis o en trans con una beta-talasemia, los valores de HbA2 se reducen a los valores normales, pudiendo cambiar el fenotipo hematológico típico de rasgo talasémico y aumentar los riesgos de un diagnóstico equivocado17,18.
Para finalizar, habría que decir que cuando se analice un cromatograma hay que pensar no solo en la posibilidad de alfa-, beta-talasemia y hemoglobinopatías estructurales, sino también en una delta-talasemia o variante de HbA2. Se deberían considerar cada uno de los picos y sus porcentajes para su correcta interpretación con el objetivo de evitar, en lo posible, diagnósticos erróneos.
FinanciaciónEste estudio ha sido financiado por la Asociación Madrileña de Hematología y Hemoterapia 2012 y la beca del FIS número PI12/01068.
AutoríaTodos los autores han tenido acceso completo a los datos, participado en el análisis y/o interpretado los resultados, y redactado el manuscrito. Todos los autores han leído y aprobado el manuscrito final.
Conflicto de interesesLos autores declaran que no tienen ningún conflicto de intereses.
