




Información del artículo
Texto completo
Bibliografía
Descargar PDF
Estadísticas
Tablas (2)
Tabla 1. Elementos de síntesis comparativa entre las diferentes visiones de la enfermedad: conceptos
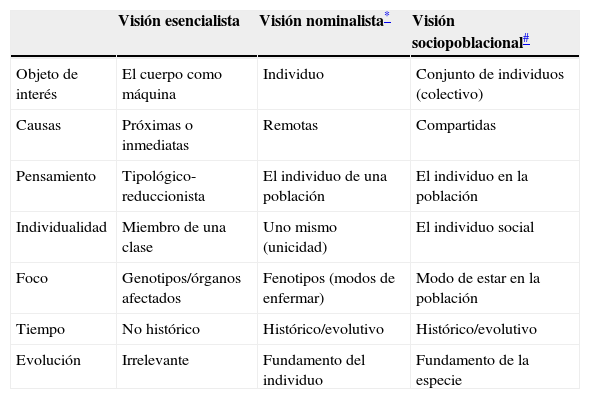
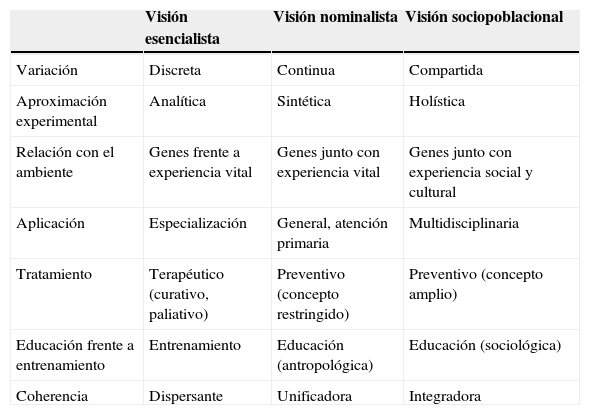
Tabla 2. Elementos de síntesis comparativa entre las diferentes visiones de la enfermedad: consecuencias
Mostrar másMostrar menos
Artículo
Opciones para acceder a los textos completos de la publicación Medicina Clínica
Suscriptor
Suscribirse

Comprar
Comprar acceso al artículo
Comprando el artículo el PDF del mismo podrá ser descargado
Precio 19,34 €
Comprar ahora
Comprando el artículo el PDF del mismo podrá ser descargado
Precio 19,34 €
Comprar ahora
Contactar
Teléfono para suscripciones e incidencias
De lunes a viernes de 9h a 18h (GMT+1) excepto los meses de julio y agosto que será de 9 a 15h
Llamadas desde España
932 415 960
Llamadas desde fuera de España
+34 932 415 960
E-mail
