








La hipertensión pulmonar tromboembólica crónica (HPTEC) es un síndrome de disnea, fatiga e intolerancia al ejercicio causado por la obstrucción tromboembólica proximal y el remodelado distal de la circulación pulmonar, lo que provoca una elevación de la presión arterial pulmonar y un fallo ventricular derecho (FVD) progresivo1. Se han postulado distintos mecanismos para explicar esta entidad, lo que incluye la recurrencia embólica después de un episodio tromboembólico pulmonar tratado adecuadamente2, la propagación in situ de un trombo en los vasos pulmonares3 y la incapacidad para disolver el émbolo inicial, lo que lleva a una vasculopatía de grandes y pequeños vasos4.
IncidenciaSu verdadera incidencia es desconocida. En una cohorte2 de 78 pacientes con tromboembolia pulmonar (TEP) aguda estudiados ecocardiográficamente, al año de seguimiento el 43,5 y el 5,1% tenían una presión arterial pulmonar sistólica (PAPs) superior a 30 y 40mmHg, respectivamente. El 75% de los pacientes con PAPs>40mmHg sufrió una endarterectomía durante los 5 años siguientes, mientras que ninguno de los que presentaron PAPs bajas necesitó cirugía. La PAPs disminuyó hasta estabilizarse aproximadamente 38 días después de la TEP aguda, aunque sin normalizarse en ningún momento. También la función ventricular derecha presentó una evolución similar. Todo ello sugiere que la realización de un ecocardiograma a las 6 semanas del episodio embólico agudo podría predecir la posible aparición de una HPTEC1.
Otro estudio3, realizado con una cohorte de 223 pacientes anticoagulados durante los 6 meses posteriores a una TEP aguda y un seguimiento aproximado de 94 meses, en el que la HPTEC fue definida como una PAPs y una presión media de la arteria pulmonar (PMAP) superior a 40 y 25mmHg, respectivamente, muestra que a los 2 años de seguimiento el 3,8% desarrolló una HPTEC. Estos 2 estudios2,3 sugieren que uno de cada 25 pacientes con un episodio inicial de TEP aguda padecerá una HPTEC.
FisiopatologíaEl tratamiento de la TEP aguda generalmente mejora la situación hemodinámica pulmonar1. Sin embargo, y a pesar de realizarse una anticoagulación adecuada, al año, los trombos residuales pueden persistir hasta en el 50% del total de los pacientes1. Si el episodio agudo no se resuelve en 1 a 4 semanas, el material embólico se incorpora a la pared del vaso, y con el tiempo es remodelado dentro del tejido conectivo y elástico, que contiene células precursoras endoteliales y musculares lisas1. Estudios histopatológicos5 y endoscópicos1 de las arterias pulmonares realizados a las pocas semanas de una TEP aguda, revelan una estenosis del vaso en el lugar de la obstrucción y una respuesta vasoconstrictora que puede llevar a una vasculopatía arteriolar de pequeño vaso caracterizada por una excesiva proliferación de células vasculares e inflamatorias alrededor de las arteriolas precapilares de la circulación pulmonar. Factores neurohormonales, incluida la endotelina-1, podrían desempeñar un papel central en la HPTEC como potente vasoconstrictor y desencadenante de los cambios microvasculares5. La hipertensión arterial pulmonar (HAP) aparecerá cuando la capacidad del resto del lecho vascular sano no pueda absorber el gasto cardíaco, ya sea debido al grado de obstrucción primaria por el material tromboembólico y el remodelado adyacente, o a la combinación de una obstrucción proximal y una vasculopatía secundaria de pequeño vaso1.
ClasificaciónSe han descrito 4 tipos de enfermedad oclusiva pulmonar basados en la localización anatómica del trombo y en la patología de la pared del vaso6.
Enfermedad tipo 1Representa alrededor del 25% de los casos de HPTEC y se caracteriza por la aparición de trombos frescos en las arterias pulmonares principales o lobares.
Enfermedad tipo 2Representa alrededor del 40% de los caso de HPTEC y se define por el engrosamiento y la fibrosis de la íntima con o sin trombos organizados próximos a las arterias segmentarias.
Enfermedad tipo 3Representa alrededor del 30% de los caso de HPTEC y se caracteriza por el engrosamiento y la fibrosis de la íntima, con o sin trombos organizados dentro de las arterias segmentarias distales y subsegmentarias.
Enfermedad tipo 4Representa algo menos del 5% de todos los casos de hipertensión pulmonar tromboembólica. Es una vasculopatía microscópica arteriolar distal sin enfermedad tromboembólica visible y es inoperable.
Factores de riesgoEn la tabla 1 se incluyen los factores de riesgo para la HPTEC, que en la mayoría de los casos no está relacionada con defectos específicos de la coagulación o con enfermedades médicas subyacentes1.
Factores de riesgo para la hipertensión pulmonar tromboembólica crónica
| Factores específicos de TEP |
| Episodios múltiples de TEP |
| Grandes defectos de perfusión |
| Pacientes jóvenes |
| PAPs superior a 50mmHg como manifestación inicial de TEP |
| Hipertensión pulmonar persistente detectada ecográficamente 6 meses después de un TEP |
| Enfermedades crónicas |
| Postesplenectomía |
| Enfermedades inflamatorias crónicas, incluidas la enfermedad inflamatoria intestinal y la osteomielitis |
| Catéteres intravenosos permanentes |
| Shunts ventriculoauriculares |
| Marcapasos infectados |
| Estomatocitosis hereditaria |
| Síndrome de Klippel-Trenaunay |
| Terapia tiroidea de sustitución |
| Cáncer |
| Factores trombóticos |
| Anticoagulante lúpico |
| Anticuerpos antifosfolípido |
| Factor viii elevado |
| Déficits de antitrombina iii, proteínas C y S |
| Disminución de los niveles de trombomodulina |
| Anticuerpos antiplaquetarios inducidos por heparina |
| Incremento de la resistencia a la fibrinólisis |
| Factores genéticos |
| Grupo sanguíneo distinto de O |
| Polimorfismos HLA |
| Fibrinólisis endógena anormal |
HLA: antígenos de leucocitos humanos; PAPs: presión arterial pulmonar sistólica; TEP: tromboembolia pulmonar.
Fuente: elaboración propia a partir de varios autores1,5.
Para que la hipertensión pulmonar se manifieste clínicamente es necesario que se obstruya entre el 60 y el 70% del lecho vascular, por lo que estos pacientes, inicialmente, suelen presentar una serie de síntomas inespecíficos7. El más frecuente es la disnea de esfuerzo progresiva con intolerancia al ejercicio, que no guarda relación con las alteraciones encontradas en la exploración física. A medida que la enfermedad evoluciona, pueden aparecer síntomas adicionales como el dolor torácico, -hasta en el 50% de los casos-, vértigos, síncopes, o hemoptisis, estas últimas provocadas por los vasos anormalmente distendidos por la presión intravascular7. La distensión abdominal, los edemas periféricos, la hepatomegalia y la ascitis pueden desarrollarse a medida que se instaura el FVD. Cuando este último progresa, pueden encontrarse signos típicos de hipertensión pulmonar, lo que incluye las grandes ondas V en el pulso venoso yugular, un ventrículo derecho palpable en el borde esternal inferior izquierdo, o un ritmo de galope ventricular derecho7.
DiagnósticoA todos los pacientes con historia clínica de trombosis venosa profunda, TEP o ambas, que presenten disnea, intolerancia al ejercicio, o evidencia clínica de FVD, se les debe descartar/confirmar una HPTEC. La evaluación diagnóstica de esta entidad tiene 3 objetivos1:
- 1.
Establecer la presencia y gravedad de la HAP y de la disfunción cardíaca resultante.
- 2.
Determinar su causa.
- 3.
Si la enfermedad tromboembólica está presente, determinar en qué grado puede ser corregida quirúrgicamente. El diagnóstico diferencial de una posible HPTEC obliga a realizar una serie de pruebas para establecer 3 criterios:
- a)
Confirmar la HAP, lo que requiere que la resistencia vascular pulmonar (RVP) en reposo, medida por cateterismo cardíaco derecho, sea >3 unidades Wood y que la PAPs, también en reposo, y la PMAP excedan los 40 y 25mmHg, respectivamente. Un ecocardiograma es útil como exploración inicial, pero insuficiente como prueba diagnóstica8.
- b)
La angiografía o la gammagrafía de ventilación/perfusión (V/Q) deben demostrar una obstrucción en cualquiera de las arterias principales, lobares, segmentarias o subsegmentarias, a pesar de que el paciente haya sido anticoagulado previamente durante 3 meses. Un angiograma pulmonar o una gammagrafía de V/Q normales excluye el diagnóstico9. Puede observarse una angio-tomografía computarizada (angio-TC) relativamente normal a pesar de que la gammagrafía de V/Q presente anomalías sustanciales. De ahí la importancia de esta última en la evaluación de la HPTEC10.
- c)
Descartar otras causas de HAP, fundamentalmente las asociadas con enfermedad cardíaca izquierda o con enfermedad pulmonar parenquimatosa. Para excluir la primera es necesario que la presión de oclusión de la arteria pulmonar sea inferior a 15mmHg11. En algunos pacientes puede ser más elevada debido a una dilatación del ventrículo derecho (VD) importante, a una dependencia interventricular, y al resultante de una disfunción ventricular izquierda. En estos casos, la resistencia vascular pulmonar (RVP) es superior a 600 dinas/s/cm−511.
- a)
Las pruebas funcionales respiratorias son necesarias para valorar la disnea y excluir la presencia de una obstrucción de las vías aéreas o de una fibrosis pulmonar. La capacidad de difusión de monóxido de carbono puede estar moderadamente reducida y el 20% de los pacientes tendrán defectos restrictivos moderados causados por las atelectasias secundarias a infartos pulmonares previos12,13.
La ecocardiografía transtorácica es utilizada como prueba inicial para observar la HAP. Los hallazgos adicionales incluyen la regurgitación tricuspídea y el crecimiento del VD sobre la cavidad ventricular izquierda con disfunción sistólica y diastólica de esta última14. La ecografía con contraste puede mostrar la apertura de una comunicación intraauricular como consecuencia del aumento de la presión auricular derecha15.
La gammagrafía pulmonar de V/Q es crítica para establecer el diagnóstico de HPTEC10. Un hallazgo normal excluye su diagnóstico, mientras que múltiples defectos bilaterales de perfusión lo sugieren5. Sin embargo, puede subestimar la magnitud de los defectos de perfusión debido a la existencia de una recanalización parcial de la luz de los vasos mientras todavía hay una obstrucción significativa del flujo sanguíneo16. Tampoco localiza anatómicamente la extensión de la enfermedad, por lo que no debe emplearse para determinar la accesibilidad quirúrgica5.
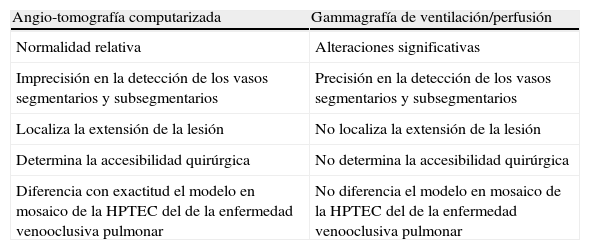
La angio-TC permite valorar la dilatación del VD y de las arterias pulmonares centrales, así como detectar los trombos organizados en las arterias principales, lobares, segmentarias y subsegmentarias, aunque en estas últimas con menor exactitud5,10. El patrón en mosaico que refleja áreas hiperperfundidas que alterna con otras hipoperfundidas también suele observarse en la enfermedad venooclusiva pulmonar, aunque en esta se asocia con engrosamientos de los septos interlobulares17. En la tabla 2 se recogen las diferencias entre la angio-TC y la gammagrafía de V/Q en el diagnóstico de la HPTEC.
Diferencias entre la angio-tomografía computarizada y la gammagrafía de ventilación/perfusión en la exploración de la hipertensión pulmonar tromboembólica crónica
| Angio-tomografía computarizada | Gammagrafía de ventilación/perfusión |
| Normalidad relativa | Alteraciones significativas |
| Imprecisión en la detección de los vasos segmentarios y subsegmentarios | Precisión en la detección de los vasos segmentarios y subsegmentarios |
| Localiza la extensión de la lesión | No localiza la extensión de la lesión |
| Determina la accesibilidad quirúrgica | No determina la accesibilidad quirúrgica |
| Diferencia con exactitud el modelo en mosaico de la HPTEC del de la enfermedad venooclusiva pulmonar | No diferencia el modelo en mosaico de la HPTEC del de la enfermedad venooclusiva pulmonar |
HPTEC: hipertensión pulmonar tromboembólica crónica.
Fuente: modificada a partir de varios autores1,5,10,17.
La angiografía pulmonar es la prueba de oro para definir la anatomía vascular pulmonar1. No obstante, pensamos que al ser un procedimiento agresivo no exento de riesgos, debería reservarse, al igual que la angioscopia pulmonar18 (con la que puede realizarse conjuntamente), para los casos en los que no exista una certeza diagnóstica de HPTEC, o para definir la ubicación exacta de la obstrucción trombótica, previamente a la realización de una endarterectomía19. Las imágenes angiográficas varían desde defectos de llenado hasta vasos completamente trombosados. El material trombótico organizado a lo largo de la pared vascular origina un borde luminal dentado o festoneado. El engrosamiento de las paredes y la dilatación de los vasos proximales provoca que su luz llena de contraste tenga un diámetro aparentemente normal20.
TratamientoEl tratamiento de elección de la HPTEC es la endarterectomía pulmonar. Esta recomendación se basa en el hecho de que es potencialmente curativa, ya que normaliza casi por completo los parámetros hemodinámicos y mejora el estado clínico del paciente1. Aquellos con RVP elevadas y obstrucción vascular mínima (enfermedad tipo 4) son los que tienen peor pronóstico, ya que la cirugía no corrige la hipertensión en esta población6,18. La vasculopatía arteriolar precapilar sin tromboembolia de los grandes vasos no es modificada por la endarterectomía de las arterias proximales. La mayoría de las muertes postoperatorias ocurren en este subgrupo, por lo que los esfuerzos realizados deben dirigirse a identificar mejor a este tipo de pacientes para evitar en lo posible operaciones innecesarias21. En la tabla 3 se recogen los factores pronósticos más importantes a tener en cuenta antes de realizar una endarterectomía pulmonar.
Endarterectomía pulmonar: factores pronósticos
| Resistencia vascular pulmonar |
| Los pacientes con RVP elevadas y obstrucción vascular mínima (enfermedad tipo 4) son los que presentan peor pronóstico |
| La vasculopatía arteriolar precapilar sin enfermedad tromboembólica de grandes vasos no es modificada por la endarterectomía de las arterias pulmonares proximales |
| La mayoría de las muertes postoperatorias se dan en este grupo de pacientes |
| Una RVP preoperatoria<1.200 dinas/s/cm−5 y la ausencia de comorbilidades graves es de buen pronóstico |
| Tipo de HPTEC |
| Los pacientes con enfermedad tipo 3 y 4 tienen una regurgitación tricuspídea, PAPs y RVP mayores que aquellos con enfermedad tipo 1 y 2 |
| Los pacientes con enfermedad tromboembólica distal (tipo 3 y 4) tienen mayor mortalidad peroperatoria, requieren más soporte inotrópico y mayor estancia hospitalaria que los del tipo 1 y 2 |
| El grado de mejoría en la HP y la regurgitación tricuspídea después de la endarterectomía es determinado por el tipo y la localización de la tromboembolia pulmonar |
HP: hipertensión pulmonar; HPTEC: hipertensión pulmonar tromboembólica crónica; PAPs: presión arterial pulmonar sistólica; RVP: resistencia vascular pulmonar.
Fuente: modificada a partir de varios autores1,21,32.
Los pacientes sometidos a cirugía, generalmente presentan una RVP preoperatoria>300 dinas/s/cm−5, a menudo con un rango de 800 a 1.400 dinas/s/cm−522. Aquellos en los que la RVP postoperatoria disminuye por lo menos un 50%, con valores menores a 500 dinas/s/cm−5, tienen mejor pronóstico que los que no lo hacen5. No hay límite superior para la RVP, ni para el grado de disfunción VD o de regurgitación tricuspídea que pueda excluir a un paciente de este tipo de cirugía en centros con la experiencia suficiente, que por desgracia son relativamente pocos en nuestro país. Las alteraciones hemodinámicas o ecocardiográficas graves no deben utilizarse para etiquetar a un paciente de inoperable23. La presencia de comorbilidades que pueden afectar a la supervivencia a corto y largo plazo debe considerarse en toda evaluación preoperatoria. La edad avanzada (>80 años), la insuficiencia renal y la disfunción hepática no son contraindicaciones absolutas para la endarterectomía1. La única contraindicación absoluta para la cirugía es la enfermedad parenquimatosa pulmonar, debido a que la endarterectomía puede mejorar los parámetros hemodinámicos, pero no revierte los síntomas y la progresión de la enfermedad pulmonar18.
Resultados a corto y a largo plazo de la endarterectomía pulmonarA corto plazo, estudios ecocardiográficos14,18 han demostrado una rápida disminución en las dimensiones de la aurícula y el VD postendarterectomía, así como una mejoría significativa de la regurgitación y de la función valvular tricuspídea, que retorna a la normalidad en unos pocos días como resultado de la restauración de la geometría anular valvular después de la remodelación del VD. Por otra parte, la recuperación del flujo sanguíneo de las regiones pulmonares previamente ocluidas provoca una reducción inmediata de la RVP, con el consiguiente incremento del gasto cardíaco1. La mortalidad peroperatoria en grandes series de pacientes recogidas en centros especializados y con experiencia quirúrgica oscila entre el 2,5 y el 4,7%1. El edema de pulmón que aparece en el 5 al 15% de los casos, y consecuencia de la reperfusión inmediata de nuevas áreas del mismo, es la complicación postendarterectomía más frecuente24.
Estudios actuales25,26 demuestran que las tasas de supervivencia a los 6 años de la endarterectomía oscilan entre el 75 y el 92%, manteniéndose los beneficios hemodinámicos, la mejoría del intercambio gaseoso, del estado funcional y de la calidad de vida. En la tabla 4 resumimos los beneficios obtenidos a corto y a largo plazo, así como las complicaciones más frecuentes postendarterectomía.
Endarterectomía pulmonar
| Resultados a corto plazo | Resultados a largo plazo | Morbilidad postoperatoria |
| Mortalidad peroperatoria: 2,5-4,7% | Supervivencia a los 6 años: 75%. Mortalidad a partir de los 6 años: 7,7-25% | Edema pulmonar: 5-15% |
| Preoperatoriamente: más del 91,3% de los pacientes son clasificados como clase funcional iii-iv de la NYHA | Un año después de la endarterectomía: el 91,4% de los pacientes son reclasificados como clase funcional i-ii de la NYHA | Hemorragia pulmonar: 3,8% |
| Desaparición de la hipertrofia y la dilatación de la aurícula y ventrículo derechos. Recuperación funcional de la válvula tricúspide. Restauración del flujo sanguíneo pulmonar de las áreas obstruidas, disminución de la RVP, e incremento del gasto cardíaco | Mejoría del intercambio gaseoso, del estado funcional y de la calidad de vida de los pacientes. El 62% recupera su trabajo habitual. El 10% necesita oxigenoterapia permanentemente | Tiempo medio operatorio: 6,7h (3,5-13,1h). Tasa de infecciones peroperatorias: 2,4% |
NYHA: New York Heart Association; RVP: resistencia vascular pulmonar.
Fuente: modificada a partir de varios autores1,14,32.
Se ha investigado la utilidad de distintos marcadores para predecir la mortalidad después de la endarterectomía pulmonar. Tanto el brain natriuretic peptide (BNP «péptido natriurético cerebral»)27 como la fracción N-terminal de su propéptido (NT-proBNP)28, las proteínas fijadoras de ácidos grasos cardíacos29, la proteína C reactiva30 y la trombomodulina31 han demostrado una buena correlación con la RVP o la función VD en pacientes operados. En la figura 1 se expone el algoritmo diagnóstico de la HPTEC.
: gammagrafía de ventilación/perfusión; TC: tomografía computarizada; TEP: tromboembolia pulmonar; TVP: trombosis venosa profunda. Fuente: elaboración propia a partir de varios autores1,5,17.")
Algoritmo diagnóstico de la hipertensión pulmonar tromboembólica crónica.
FVD: fallo ventricular derecho; HPTEC: hipertensión pulmonar tromboembólica crónica; Gammagrafía (V/Q): gammagrafía de ventilación/perfusión; TC: tomografía computarizada; TEP: tromboembolia pulmonar; TVP: trombosis venosa profunda.
Fuente: elaboración propia a partir de varios autores1,5,17.
Todos los pacientes con HPTEC objetivamente demostrada, y en ausencia de contraindicaciones, sean o no candidatos a tratamiento quirúrgico, deben anticoagularse indefinidamente con las dosis adecuadas para mantener una ratio internacional normalizada entre 2 y 3, tanto antes como después de la endarterectomía32. Sin embargo, este tratamiento se acompaña de una mortalidad a los 3 años del 90%33. Para tratar de reducirla se han añadido a la anticoagulación una serie de medicamentos conocidos genéricamente como terapia médica específica para la HAP (terapia específica-HAP), que incluye los antagonistas de los receptores de la endotelina (bosentan), los inhibidores de la fosfodiesterasa (sildenafilo) y los análogos de la prostaciclina (epoprostenol y treprostinil)34. Solo disponemos de un ensayo clínico multicéntrico, aleatorio y controlado con placebo para la terapia específica-HAP en la HPTEC, el BENEFIT35(Bosentan Effects in Inoperable Forms of Chronic Thromboembolic Pulmonary Hypertension), que evalúa el bosentan. Este estudio, en el que participaron 157 pacientes con HPTEC inoperable o enfermedad residual posquirúrgica, muestra a las 16 semanas de tratamiento una reducción muy modesta de la RVP, en cualquier caso siempre inferior a la experimentada por los pacientes endarterectomizados23. Tampoco hubo mejoría en la capacidad funcional ni en la prueba de la marcha de 6 minutos. No existen referencias sobre la terapia específica-HAP que demuestren claramente un beneficio en términos de síntomas, capacidad de ejercicio o supervivencia. Las informaciones sobre este tipo de tratamiento provienen de evaluaciones retrospectivas realizadas sobre series pequeñas y sin controles de distintos fármacos1. Estos datos no definen si una clase de fármaco es superior a las otras, o si la terapia combinada es una aproximación racional. No se ha demostrado que la terapia específica-HAP aumente la supervivencia, ya sea como medicación única o en conjunción con la endarterectomía7.
Con todo ello podemos establecer que todo paciente con antecedentes de trombosis venosa profunda y/o TEP, con disnea, intolerancia al ejercicio, o evidencia clínica de FVD debe ser evaluado para descartar/confirmar una HPTEC. En todo paciente que haya padecido una TEP aguda es aconsejable realizar un ecocardiograma 6 semanas después para confirmar una HAP persistente, ya que puede predecir el desarrollo de una HPTEC. El tratamiento de elección en todo paciente diagnosticado objetivamente de HPTEC es la endarterectomía.
En ausencia de contraindicaciones, todo paciente diagnosticado objetivamente de HPTEC debería anticoagularse indefinidamente. Solo se recomienda la terapia específica-HAP en pacientes con HPTEC considerados inoperables debido a comorbilidades graves, o por propia elección del enfermo, así como en aquellos con HAP residual postendarterectomía que no están en condiciones de repetirla en un centro con más experiencia.
La terapia específica-HAP no debe utilizarse en lugar de ni como tratamiento puente de la endarterectomía pulmonar, ya que lo único que se consigue es aplazar esta, con el riesgo consiguiente para el paciente.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Los autores expresan su agradecimiento a RETIC COMBIOMED (RDO7/0067/0006).
