







La inclusión del año en curso en el título de esta revisión puede parecer una exageración, pero con ello se pretende resaltar la enorme velocidad con la que están cambiando los conceptos en el mieloma múltiple (MM). Aunque su definición como neoplasia derivada de células de estirpe linfoide B en el último estadio madurativo capaces de sintetizar inmunoglobulinas (Ig) (células plasmáticas) se mantiene vigente desde sus primeras descripciones, pocas enfermedades se han visto sometidas a un cambio tan radical en los últimos años. Desde sus criterios diagnósticos hasta las posibilidades terapéuticas, pasando por la patogenia, el manejo de las complicaciones o los criterios de respuesta, prácticamente todos estos aspectos se han modificado en la última década. En esta revisión abordaremos una descripción clásica del MM, pero centrada en los aspectos que por su novedad necesitan una mayor actualización.
Epidemiología
El MM constituye, por frecuencia, la segunda neoplasia hematológica tras el linfoma. Según datos de 2006 en EE.UU.1, su incidencia es de 56 casos nuevos por cada 1.000.000 de habitantes y año. Además, se sitúa como la décima causa de muerte por cáncer en números absolutos (3,8 muertes por cada 100.000 habitantes y año). La edad de aparición se sitúa en los 69 años y sólo un 15% de los casos se diagnostica antes de los 50 años. No hay diferencias relevantes en la incidencia por sexos y, respecto a la raza, se presume una mayor incidencia en negros americanos y población caribeña respecto a otras poblaciones, pero estas diferencias no están totalmente aclaradas.
Manifestaciones clínicas
El dolor óseo es el síntoma más típico y frecuente. Aparece en el 60-80% de los pacientes en el momento del diagnóstico, generalmente en la columna vertebral, pero también en el esternón, las costillas o la zona proximal de las extremidades2. Su origen son lesiones óseas que, por radiología, suelen corresponder a típicas imágenes osteolíticas en «sacabocados» sin esclerosis periférica, pero también pueden manifestarse como osteoporosis grave o fracturas patológicas. Las osteólisis son incluso más frecuentes que el dolor, pues aparecen en el 80% de los casos y en el resto hay lesiones microscópicas que, aun siendo invisibles en la radiografía convencional, pueden dar imágenes visibles por resonancia magnética (RM). Se localizan sobre todo en huesos con gran cantidad de médula ósea, es decir, por orden de frecuencia: cráneo, columna vertebral, costillas, pelvis y epífisis proximales de huesos largos. El origen de estas lesiones hay que buscarlo en un desequilibrio entre resorción y formación óseas. En etapas iniciales se observa un aumento de la resorción por activación de osteoclastos debida al aumento de estimulantes como el factor de necrosis tumoral alfa y beta, la interleucina (IL) 1-ß e IL-6, pero sobre todo a una hiperactividad del receptor del activador del factor nuclear kappa y su ligando (RANK/RANK-L), ya que el ligando se expresa en células plasmáticas y estroma2. El aumento destructivo se compensa al principio con un aumento de la formación ósea, pero al crecer el clon tumoral se sintetiza Dickkopf-1 (DKK1), que es un factor que inhibe la vía de Wingless tipo-1 (WNT1), esencial en los osteoblastos3. Así pues, la formación ósea se resiente y se acaba por generar un desequilibrio que ocasiona una pérdida neta de masa ósea, lo que genera la clásica tríada de la lesión ósea del mieloma: dolor, hipercalcemia y fracturas patológicas. La hipercalcemia está presente en un tercio de los pacientes en el momento del diagnóstico y un 30% adicional la desarrolla durante la evolución de la enfermedad. Las fracturas patológicas más frecuentes aparecen en la columna vertebral y pueden motivar lesiones radiculares y/o medulares, aunque en estos casos suele haber un plasmocitoma asociado.
Otra manifestación clínica clásica del mieloma es la anemia. Hasta el 50% de los pacientes presenta anemia moderada (aproximadamente 10 g/dl) y un 25% de los casos, anemia grave (< 8 g/dl)4. Como en tantas enfermedades neoplásicas, su causa es multifactorial, si bien los mecanismos más importantes son 2: a) reducción de la concentración de eritropoyetina circulante, debido al aumento de citocinas como el factor de necrosis tumoral alfa e IL-1, y a la posible existencia de lesión renal asociada, y b) presencia del ligando de Fas en la superficie de las células plasmáticas, que, al activar a Fas en los progenitores eritroides, activa la apoptosis5. Otras causas de anemia pueden ser la invasión masiva de la médula ósea, la quimioterapia, la presencia de déficit asociados de vitamina B12 y/o folato, y el efecto diluyente de la hiperproteinemia plasmática. Junto a la anemia, también son frecuentes los síntomas constitucionales (astenia y pérdida de peso).
La insuficiencia renal (IR) aparece en un 25-30% de los pacientes, aunque las series más recientes describen una frecuencia menor6,7. Su origen es multifactorial, si bien la causa más descrita es la eliminación renal de cadenas ligeras de Ig (proteinuria de Bence-Jones), cuya presencia en los túbulos se denomina histológicamente «riñón de mieloma». Otras causas son hipercalcemia, hiperuricemia, deshidratación, infecciones urinarias de repetición, hiperviscosidad, amiloidosis e infiltración tumoral. Suele ser una IR crónica, pero también puede manifestarse como IR aguda (más raramente como síndrome de Fanconi adquirido o síndrome nefrótico).
Las infecciones son la primera causa de mortalidad del MM, con una incidencia de 0,8-2,2 episodios por paciente y año, entre 7 y 15 veces por encima de la observada en pacientes hospitalizados por otros motivos6. Suelen ser bacterianas y localizadas en el pulmón y las vías urinarias, pero también las hay virales (sobre todo, herpes zóster) y fúngicas. La causa principal de la susceptibilidad infecciosa es la hipogammaglobulinemia policlonal, pero también hay otros motivos: reducción de células T CD4+, alteración funcional de las células citolíticas, anomalías en el sistema del complemento y granulopenia ocasional.
El síndrome de hiperviscosidad es raro (4-8%) en el MM. Su génesis se relaciona con la concentración de la paraproteína y con las características moleculares de ésta (es más frecuente con IgM excepcional en el MM, IgG3 e IgA)8. Hasta el 10-15% de los mielomas presentan amiloidosis, aunque con poca participación orgánica. Otras manifestaciones clínicas son diátesis hemorrágica, hepatomegalia (un 20% de los casos), esplenomegalia (< 10%) y plasmocitomas (tumoraciones de células plamáticas) en cualquier localización.
Exploraciones complementarias
Tras una sospecha clínica o como hallazgo casual, la investigación de un paciente con MM requiere una serie de exploraciones complementarias obligadas para confirmar el diagnóstico, establecer el pronóstico y orientar el tratamiento. En la tabla 1 se exponen las exploraciones necesarias según el momento diagnóstico concreto del paciente, tal como se presentan en la guía británica9. De todas ellas, destacamos a continuación los datos más relevantes.
Parámetros electroforéticos
Proteinograma. Permite identificar el marcador biológico más característico del mieloma: la presencia en suero y/u orina de una Ig monoclonal (paraproteína o componente monoclonal). El isotipo es IgG en el 60% de los casos; IgA en el 25-30%, e IgD en el 2%10. Los mielomas IgE e IgM son excepcionales. El resto (10-15%) carece de cadena pesada y sólo tiene cadenas ligeras MM Bence-Jones (BJ). La cantidad de componente monoclonal se ha utilizado para distinguir entre MM y gammapatía monoclonal de significado incierto (GMSI), pero ya no se emplea con este fin. No obstante, sí es útil para el seguimiento de la evolución de la enfermedad. El valor de albúmina suele estar reducido, hecho que se asocia a mal pronóstico. Por último, la concentración de Ig policlonales está disminuida en mielomas sintomáticos, a diferencia de lo que sucede en casos indolentes y GMSI.
El método de elección para llevar a cabo el proteinograma es la electroforesis en agarosa o acetato de celulosa, aunque tiene poco valor para evaluar las cadenas ligeras y paraproteínas tipo IgD. Para ello se requiere una técnica más sensible, como la electroforesis en gel de agarosa de alta resolución. El MM IgD y el BJ deben sospecharse si no se identifica un aumento de alguna Ig pese a existir con un pico monoclonal o si hay hipogammaglobulinemia intensa. Una vez que se ha detectado el componente monoclonal, se debe identificar el isotipo de las cadenas ligera y pesada por inmunoelectroforesis o inmunofijación (IFE). La IFE es más sensible y por ello la más usada para detectar componentes monoclonales residuales tras el tratamiento. El isoinmunoelectroenfoque es un método alternativo de alta sensibilidad para detectar cantidades muy pequeñas, pero requiere personal altamente cualificado y muestra diagnóstica de referencia11. La nefelometría se ha usado con frecuencia para detectar el tipo de cadena ligera monoclonal, basado en la existencia de un desequilibrio en el cociente kappa/lambda. Sin embargo, hay casos con baja secreción que presentan un cociente kappa/lambda normal. En estos casos se impone la utilización de la IFE o la cuantificación de cadenas ligeras libres (véase más adelante).
Detección, identificación y cuantificación de proteínas monoclonales. El método de elección es la nefelometría, siendo inaceptable la turbidimetría. La cantidad de Ig policlonales está disminuida (inmunoparesia) en el 75% de los mielomas. La cuantificación de Ig también sirve para evaluar cambios en el componente monoclonal, parámetro de gran utilidad en el seguimiento de la respuesta al tratamiento, aunque para esta evaluación es más recomendable basarse en los cambios del componente monoclonal por electroforesis. No obstante, lo más importante es seleccionar una de las 2 técnicas y usar siempre la misma durante el curso de la enfermedad. En caso de buenas respuestas al tratamiento, se requiere usar la IFE para confirmar la completa desaparición del componente monoclonal. La inmunosustracción permite identificar el isotipo del componente monoclonal con gran exactitud, pero su sensibilidad es inferior a la de la IFE.
Análisis de orina. La evaluación urinaria de la proteína M requiere recoger la orina de 24 h para determinar la cantidad diaria excretada. Después la orina debe ser concentrada 200 veces (2-4 g/dl) para efectuar la electroforesis. Si la filtración de cadenas ligeras es muy abundante, se supera la capacidad de reabsorción y catabolismo de los túbulos renales y las cadenas aparecen en la orina, detectándose como una banda densa en el acetato de celulosa o un pico en el densitómetro. Esto se denomina proteinuria de BJ, y aparece en todos los mielomas BJ y hasta en el 60% del resto de los MM. Para definir el tipo de cadena ligera monoclonal se debe usar orina concentrada con anticuerpos frente a kappa y frente a lambda. En caso de cantidades pequeñas de proteinuria es obligatorio usar la IFE.
Detección de cadenas ligeras libres en suero. Desde hace poco se dispone de un nuevo método serum free light chain (FLC), Freelite©, The Binding Site, Birmingham, Reino Unido que utiliza anticuerpos monoclonales que detectan cadenas ligeras libres de forma específica (no se unen a cadenas ligeras ligadas a cadenas pesadas)12. Aunque todavía se requiere experiencia con esta técnica, es útil en el diagnóstico y seguimiento de mielomas BJ y no secretores, amiloidosis primaria e incluso GMSI13-15. Este método es posiblemente más sensible que la medición urinaria, porque el riñón puede ejercer cierto grado de catabolismo proteico. Por ello en algunos centros la detección sérica de cadenas ligeras libres está sustituyendo a las pruebas urinarias. También podría servir para la detección precoz de recaídas en pacientes con MM o amiloidosis, pero es algo que todavía debe confirmarse en estudios prospectivos.
Parámetros hematimétricos y de médula ósea
La anemia es frecuente (> 50%), pero la neutropenia y la trombocitopenia son raras (< 15%), salvo en estadios finales16. La velocidad de sedimentación globular está elevada (> 50 mm/h) en el 65% de los pacientes, y en el frotis se observan hematíes apilados (fenómeno de rouleaux). Dado que este fenómeno se debe a la presencia de hipergammaglobulinemia sérica, no es extraño que hasta el 15% de los mielomas (BJ, no secretores) carezcan de este hallazgo y tengan valores de velocidad de sedimentación globular menores de 20 mm/h. La presencia de plasmocitos circulantes es rara (< 15%) y se asocia con enfermedad avanzada, pero con técnicas de alta sensibilidad (citometría o reacción en cadena de la polimerasa) se detectan ciertas concentraciones de células plasmáticas circulantes en aproximadamente el 70% de los pacientes. Si el porcentaje de plasmocitos circulantes supera el 20% en número relativo y 2 * 109 células/l en número absoluto, se cumplen criterios de leucemia de células plasmáticas17.
El examen citomorfológico de la médula ósea es obligado para diagnosticar un MM, ya que es necesario demostrar la presencia de plasmocitosis. El infiltrado, de acuerdo con los criterios más extendidos, suele ser superior al 10% (promedio de aproximadamente el 50%), pero cifras inferiores no descartan el diagnóstico si se demuestra la presencia de células plasmáticas monoclonales. Su morfología es heterogénea, debido a la posible existencia de diferentes grados de maduración.
La biopsia de médula ósea se considera obligatoria en muchos centros, ya que refleja el grado de infiltración tumoral mejor que el aspirado, además de proporcionar información pronóstica. No obstante, en España suele restringirse a pacientes jóvenes o que plantean dudas diagnósticas.
Parámetros bioquímicos
Parámetros convencionales. La IR (creatinina > 2 mg/dl) es frecuente (véase más arriba) y es el parámetro que más condiciona el futuro del paciente. La hipercalcemia se detecta en un 25-30% de los pacientes, mientras que la hiperuricemia es más rara (< 15%). El valor de lactatodeshidrogenasa refleja el recambio celular y se incrementa en formas muy agresivas (en torno al 20% de los casos)6,16. El incremento de la beta-2-microglobulina es indicativo de gran carga tumoral y/o IR, y constituye uno de los principales factores pronósticos de la enfermedad (véase más adelante)18. La hipoalbuminemia es también un factor de mal pronóstico18.
Citocinas y reactantes de fase aguda. La IL-6, al actuar sobre el receptor soluble de la IL-6 (sIL-6R), induce diferenciación de linfocitos B a células secretoras y proliferación de células plasmáticas. Los valores séricos de ambos están aumentados en el 30-50% de los MM, con una cifra superior a las GMSI. Al haber casos solapados, su valor diagnóstico es relativo, pero no su valor pronóstico, ya que si los valores están altos la enfermedad suele ser más agresiva. La proteína C reactiva, reactante de fase aguda relacionado con la IL-6, está aumentada en el 40% de los MM. Un valor elevado de proteína C reactiva es un factor pronóstico adverso independiente. Puede emplearse como alternativa a la determinación de IL-6.
Estudios especiales
Inmunofenotipo. Supone una notable ayuda en el diagnóstico, pronóstico y seguimiento terapéutico. En el MM, la citometría de flujo es muy superior a otras técnicas como la inmunofluorescencia y la inmunofosfatasa alcalina. Las células plasmáticas se identifican por absorber y dispersar la luz de forma característica, por contener Ig citoplásmicas y por la expresión antigénica de CD138 (específico de células plasmáticas) y CD38 (con sobreexpresión específica de células plasmáticas)19. La distinción entre célula plasmática normal o neoplásica se establece al detectar una expresión restringida de la cadena ligera de las Ig o por la presencia de anomalías fenotípicas como ausencia de CD19, presencia de CD56, CD13, CD33 o CD117, hechos impropios de la célula plasmática normal19. Esto ayuda en el diagnóstico diferencial, ya que las GMSI tienen más de un 5% de células plasmáticas normales en la médula ósea con respecto a la celularidad plasmocitaria total, mientras que en los MM las células plasmáticas son anormales en su totalidad.
La presencia o ausencia de ciertos antígenos se ha relacionado con el pronóstico. Así, los MM CD20+ y CD45+ parecen tener un peor pronóstico, mientras que la ausencia de CD56 con expresión de CD44 se asocia a enfermedad extramedular. La expresión de CD28 se relaciona con la actividad de la enfermedad y sólo aparece en formas muy agresivas. Finalmente, la presencia de CD117 parece asociarse a un pronóstico favorable19. En cualquier caso, la mayor utilidad del inmunofenotipo es su aplicación en el estudio de la enfermedad mínima residual, ya que su sensibilidad y rapidez permiten detectar pequeñas cantidades de células, como ocurre tras el tratamiento. Así, la presencia de más de un 0,01% de células tumorales medulares y la falta de recuperación de plasmocitos normales se asocian con una mayor probabilidad de recaída en MM tras trasplante de progenitores hematopoyéticos20.
Estudios de contenido de ADN. La medida del ADN por citometría de flujo permite distinguir anomalías en el contenido de ADN de poblaciones celulares. Con ello se sabe que hasta el 55-60% de los mielomas presentan anomalías clonales en el contenido de ADN (aneuploidías), generalmente hiperploidías. Este aspecto se ha revisado recientemente, ya que se presume que hay 2 posibles vías patogénicas en el MM: una vía hiperploide frente a otra diploide o hipodiploide. En el primer caso, se generarían clones menos agresivos y, con ello, mielomas de mejor pronóstico. Estas anomalías están ya presentes desde las primeras fases de la enfermedad, pues estos hallazgos se observan ya en las GMSI, donde el porcentaje de casos aneuploides es incluso mayor (hasta el 73%)21.
Aparte de la utilidad diagnóstica reseñada, la aneuploidía de ADN puede usarse en estudios de enfermedad mínima residual, donde tiene una sensibilidad de 104, similar a la del fenotipo, aunque con menor aplicabilidad (en torno al 50%). Sin embargo, el mayor valor de la medida del ADN es su capacidad pronóstica, ya que permite determinar el porcentaje de células plasmáticas en las diferentes fases del ciclo celular. El porcentaje de células plasmáticas en fase de síntesis o fase S (con cantidad de ADN mayor de 2 n y menor de 4 n) es un parámetro directamente relacionado con la actividad proliferativa del clon tumoral, y es por ello uno de los principales factores pronósticos en el MM. Así, los pacientes con mayor porcentaje de células plasmáticas en fase S (p. ej., un 3%) tienen una supervivencia más corta que el resto22.
Citogenética y biología molecular. Inicialmente se creía que sólo el 30% de los MM presentaba alteraciones citogenéticas, pero gracias a la hibridación in situ y a las técnicas moleculares hoy se sabe que este porcentaje es de aproximadamente el 60%19,21. Las anomalías más frecuentes son las translocaciones del cromosoma 14q32 (en torno al 40% de los pacientes), que provocan la yuxtaposición del gen de la cadena pesada de las Ig frente a diversos genes. De ellos, los 5 más frecuentes son: a) CCND1 en la t(11;14)(q13;q32), presente en el 20% de los MM; b) FGFR/MMSET en la t(4;14)(p16;q32), en el 15%; c) CMAF en la t (14;16)(q32;q23), en el 5-10%; d) CCND3 en la t(6;14)(p21;q32), en el 3%, y e) MAFB en la t(14;20)(q32;q11), que es una translocación bastante rara.
La segunda anomalía más frecuente es la ganancia del brazo largo del cromosoma 1, que se observa hasta en el 50% de los casos y se asocia con frecuencia a la siguiente anomalía en orden de frecuencia: la deleción del cromosoma 13. Ésta aparece en el 30-40% de los casos cuando se hace por técnicas de hibridación in situ con sondas para el gen RB23. La presencia de esa alteración implica mal pronóstico, si bien este concepto está hoy en revisión, ya que el mal pronóstico parece deberse más a la presencia de otras anomalías asociadas que a la deleción por sí misma. También es importante por su valor pronóstico adverso la deleción de p53. Otras alteraciones son ya poco frecuentes (< 5%) y muy heterogéneas, aunque hay que señalar que la presencia de un cariotipo aberrante o hipodiploide se asocia a mal pronóstico24. En resumen, las alteraciones citogenéticas son muy frecuentes en el MM y claves en el pronóstico, por lo que se considera un estudio obligado en todo paciente con MM.
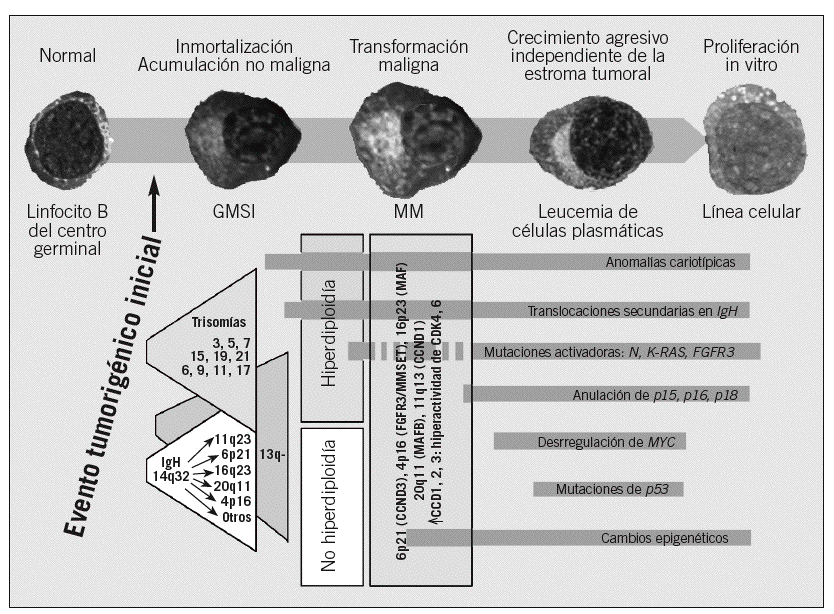
Desde el punto de vista molecular, el hallazgo más específico es el reordenamiento monoclonal de los genes de las Ig. Otras anomalías son alteraciones en C-MYC, RB, N-RAS, P53, P16, P15 y genes de ciclinas. Estas anomalías todavía no tienen una traducción clínica o patogénica confirmada, pero se ha llegado a plantear que la sobreexpresión de alguna de las ciclinas (CCND1, CCND2 o CCND3) es el hallazgo oncogénico universal en el MM (fig. 1)25, aunque esta afirmación tiene todavía que ser ratificada.
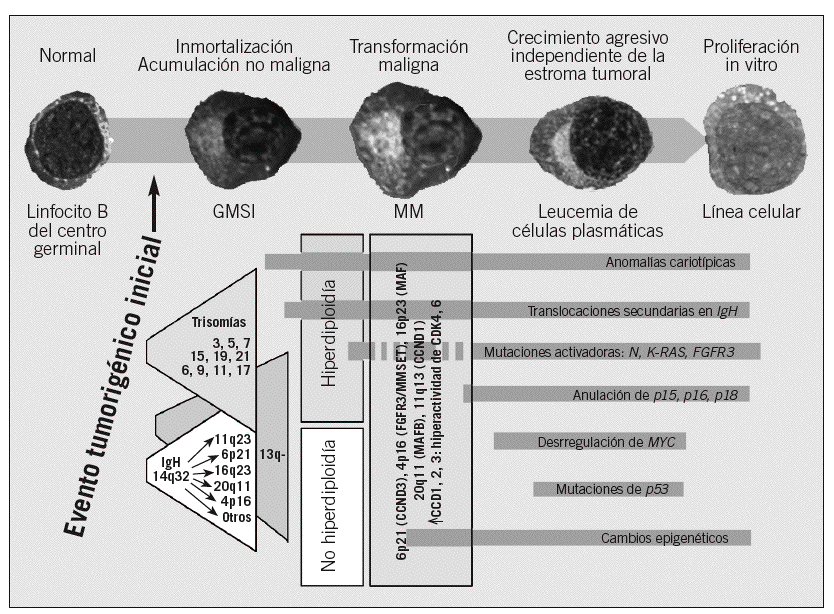
Fig. 1. Estadios de enfermedad y desarrollo secuencial de alteraciones oncogénicas. Las primeras alteraciones oncogénicas se presentan en la gammapatía monoclonal de significado incierto (GMSI) y afectan a 2 posibles vías patogénicas sin apenas solapamiento: a) translocaciones primarias en IgH, y b) trisomías múltiples. Cada una de ellas puede desarrollar también la vía patogénica de la pérdida del cromosoma 13. Otras anomalías cariotípicas, translocaciones secundarias de IgH incluidas y los cambios epigenéticos pueden aparecer en cualquier estadio. Finalmente, las mutaciones activadoras de K- o N-RAS parecen marcar, si no causar, la transición de GMSI a mieloma múltiple (MM) en algunos casos, pero también pueden producirse durante la progresión a MM. Las alteraciones oncogénicas tardías se producen cuando el clon tumoral se vuelve más agresivo, y entre ellas figuran la alteración de la regulación de MYC por translocaciones secundarias en IgH, deleción bialélica de p18, inactivación de RB y pérdida o mutación de p53. (Adaptada de Kuehl y Bergsagel. ASH educational Book, 2005. Disponible en: http://www.asheducationbook.org/cgi/content/abstract/2005/1/346).
Estudios de imagen
Radiografía convencional. Los estudios radiológicos que llevan a la sospecha de un MM suelen ser derivados de alguna semiología concreta como dolor localizado o fracturas patológicas. No obstante, cuando hay una sospecha, se debe hacer una serie ósea radiológica completa, que incluye (al menos) 13 radiografías: anteroposterior y lateral de las 3 regiones de la columna vertebral (cervical, dorsal y lumbar), anteroposterior de tórax (parrilla costal), pelvis, ambos húmeros y ambos fémures (completos) y lateral de cráneo. Se debe buscar cuidadosamente tanto las típicas lesiones osteolíticas en sacabocado como las destrucciones parciales o insuflaciones características de los plasmocitomas óseos9.
Tomografía computarizada, resonancia magnética y tomografía por emisión de positrones. Hasta ahora estas exploraciones sólo estaban justificadas en casos concretos, como lesiones osteolíticas de cabeza femoral, lesiones sacras de difícil visualización, sospecha de plasmocitomas, etc.9. Sin embargo, ya hay estudios que demuestran que tanto la RM como la tomografía por emisión de positrones pueden identificar las lesiones óseas con más precisión que la radiografía convencional. Así, permiten identificar alteraciones óseas en el 96% de los pacientes con MM (algo de gran ayuda para distinguirlo de la GMSI) y mejorar el criterio de respuesta completa, ya que pueden identificar lesiones activas residuales en pacientes en los que se considera que se ha obtenido respuesta completa con criterios actuales.
Diagnóstico
Criterios diagnósticos
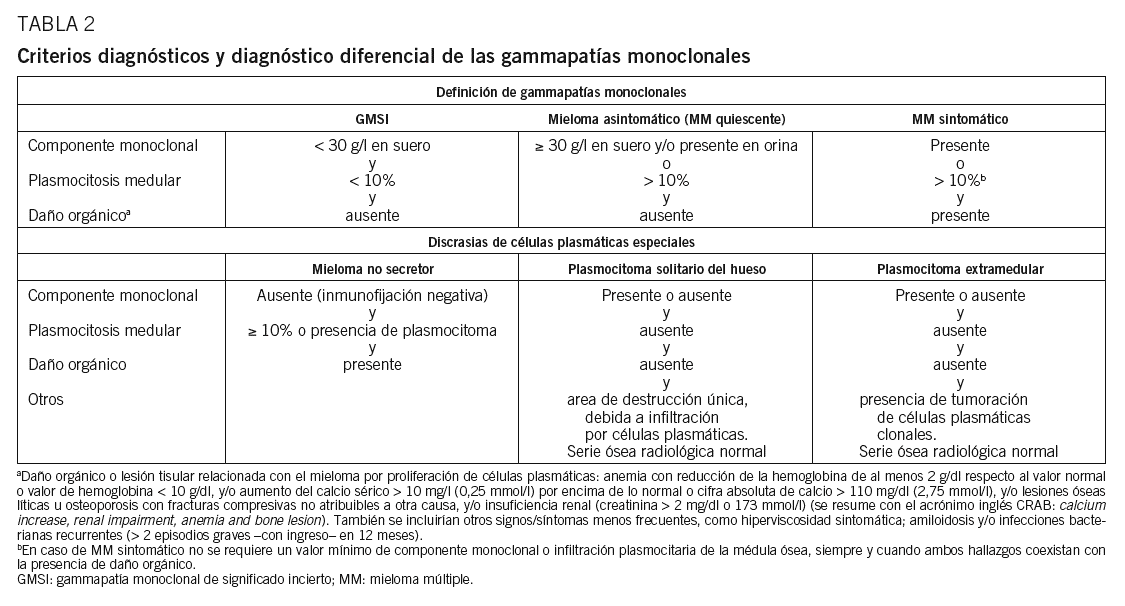
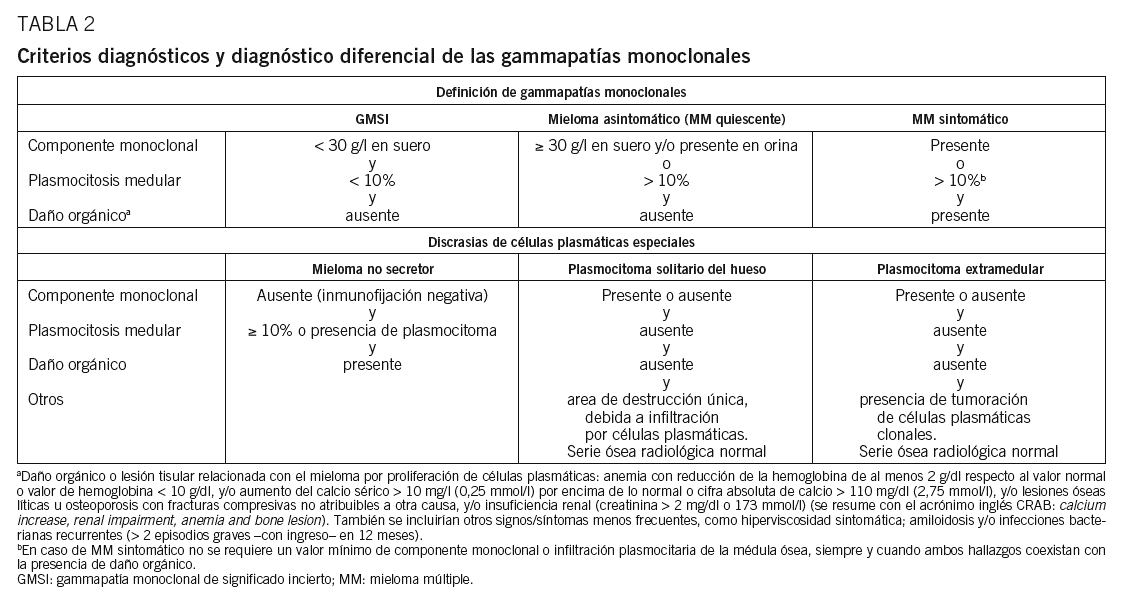
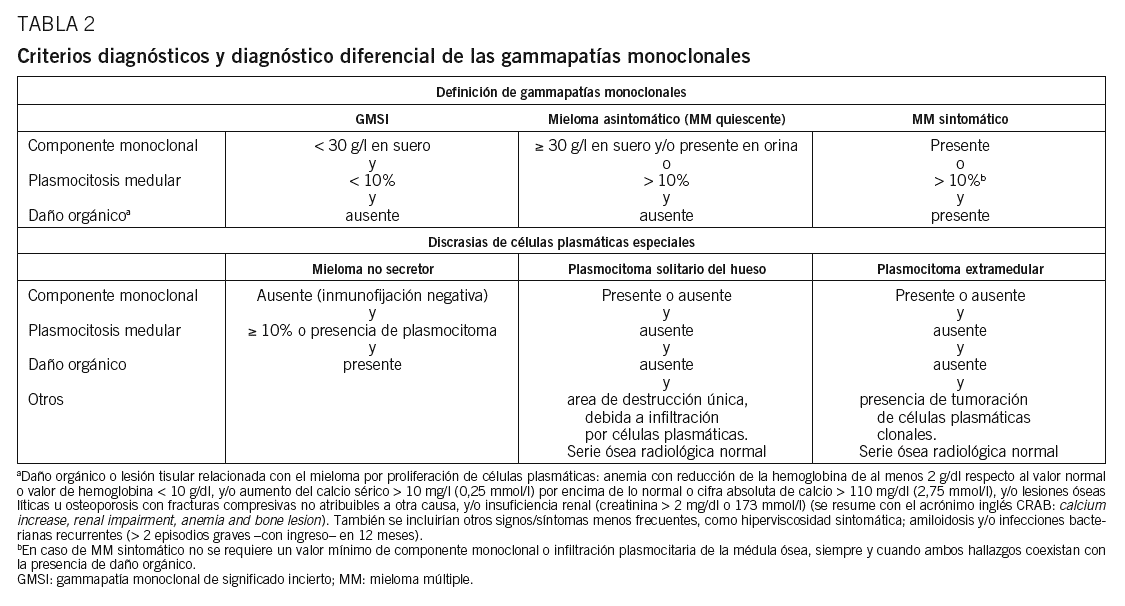
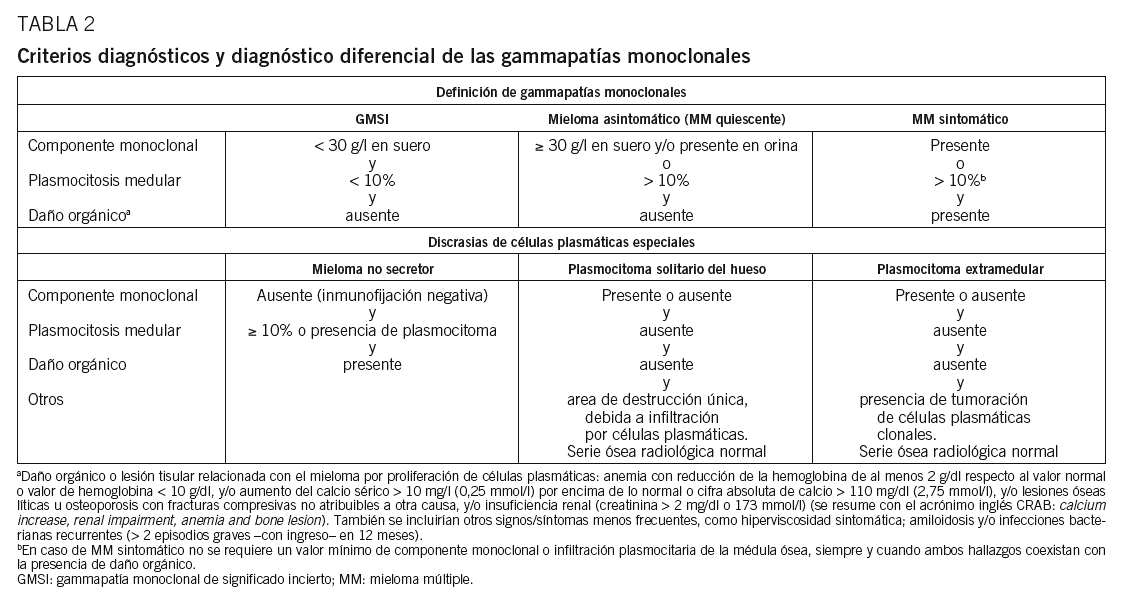
Hasta hace poco la heterogeneidad clínica del MM podía generar confusión en el diagnóstico definitivo, ya que, aunque el diagnóstico es generalmente sencillo, no es raro encontrar casos cerca del difuso límite que hay entre el MM y otras gammapatías (GMSI, amiloidosis, etc.). De hecho, había hasta 3 propuestas de criterios diagnósticos Southwest Oncology Group (SWOG), British Columbia Cancer Agency (BBCA) y Chronic-Leukemia-Myeloma Task Force, hecho que daba lugar a la paradoja de que un paciente podía tener 2 diagnósticos diferentes dependiendo de qué sistema diagnóstico se utilizara. Este problema quedó aparentemente resuelto en 2003, cuando el grupo internacional de mieloma publicó(tabla 2)26. Lo más novedoso es que el diagnóstico de un MM sintomático requiere la presencia de lesión orgánica o tisular relacionada con la neoplasia proliferante, definida por la presencia de al menos una de 7 manifestaciones posibles: a) anemia, con reducción de la hemoglobina de al menos 2 g/dl respecto al valor normal o valor de hemoglobina inferior a 10 g/dl; b) hipercalcemia mayor de 10 mg/l (0,25 mmol/l) por encima de lo normal o cifra absoluta de calcemia superior a 110 mg/dl (2,75 mmol/l); c) lesiones osteolíticas u osteoporosis con fracturas compresivas no atribuibles a otra causa; d) IR, con creatinina superior a 2 mg/dl o a 173 mmol/l (éstas son las más frecuentes y se resumen con el acrónimo inglés CRAB, de calcium increase, renal impairment, anemia y bone lesion); e) hiperviscosidad sintomática; f) amiloidosis, y g) infecciones bacterianas recurrentes (> 2 episodios graves con ingreso hospitalario en 12 meses). La presencia de alguna de estas anomalías es clave, ya que las formas sin lesión orgánica (GMSI y MM quiescente) no requieren tratamiento.
Formas clínicas especiales
Se denominan así las discrasias de células plasmáticas que no encajan en los epígrafes anteriores y deben distinguirse porque tanto la estrategia terapéutica como su seguimiento son diferentes.
Mieloma no secretor. Mieloma que carece de componente monoclonal detectable. Puede ser no productor, con células plasmáticas negativas para Ig citoplásmica, o no secretor, con síntesis pero sin secreción de Ig (células plasmáticas positivas para Ig citoplásmica). Representan aproximadamente el 1% de los MM y se diagnostican porque tienen otros criterios. Además, el cociente sérico de FLC suele ser anormal15.
Mieloma evolving. Cumple criterios de MM quiescente o GMSI, pero tiene un aumento progresivo del componente monoclonal durante su evolución. Debe diferenciarse del no evolving, ya que el primero evoluciona a MM sintomático con mucha mayor rapidez27.
Plasmocitoma óseo solitario. Tumoración de células plasmáticas localizada en una región ósea (columna, esternón, parrilla costal, huesos largos), sin infiltración de médula ósea. La supervivencia suele ser larga (> 10 años), pero un tercio de los casos evoluciona a mieloma sintomático28.
Plasmocitoma extramedular. Tumoración de células plasmáticas fuera del esqueleto, generalmente el aparato respiratorio o el gastroduodenal28.
Mieloma osteosclerótico. Forma excepcional con imágenes de osteosclerosis únicas o múltiples. Con frecuencia se acompaña de polineuropatía y componente monoclonal lambda. Cuando hay asociación de polineuropatía, lesiones osteoscleróticas, endocrinopatía, componente monoclonal sérico y lesiones cutáneas (skin) se denomina síndrome de POEMS29.
Leucemia de células plasmáticas. Presencia en sangre periférica de células plasmáticas en un porcentaje superior al 20% y, en número absoluto, por encima de 2.000/µl. En el 60% de los casos el cuadro es primario, mientras que en los restantes corresponde a la fase terminal de un MM; en ambas formas el pronóstico es malo17.
Pronóstico
El pronóstico del MM ha sufrido notables modificaciones en los últimos años, tanto en lo que se refiere a la mejora de la supervivencia (de 28 meses en la década de los ochenta se ha pasado a los 36-42 meses que se publican actualmente) como en su forma de predicción. En una revisión reciente nuestro grupo encontró más de 60 parámetros relacionados con el pronóstico, pero tal cantidad de factores tiene poca utilidad clínica y sólo sirve para evaluar ciertos mecanismos biológicos o comportamientos clínicos concretos30. Así, para tener una evaluación pronóstica con valor práctico, es decir, para orientar el tratamiento y permitir comparaciones entre distintas estrategias terapéuticas, hay que recurrir a sistemas más universales. Por fortuna, el pasado año se publicó el nuevo International Staging System (ISS)18, análogo al Índice Pronóstico Internacional de los linfomas. Se obtuvo a partir de los datos de más de 11.000 pacientes incluidos en ensayos clínicos o tratados en centros de referencia. De acuerdo con dicho índice, los pacientes se dividen en 3 grupos de riesgo: a) riesgo bajo, con valores de beta-2-microglobulina iguales o menores de 3,5 mg/l y albúmina de 3,5 mg/dl o superior; b) riesgo intermedio, con beta-2-microglobulina de 3,5 mg/l o menor y albúmina inferior a 3,5 mg/dl o beta-2-microglobulina de 3,5-5,5 mg/l, y c) riesgo alto, con beta-2-microglobulina mayor de 5,5 (tabla 3). De acuerdo con ello, la mediana de supervivencia es de 29; 44, y 62 meses, respectivamente. Aparte de estos 2 parámetros, sólo la edad puede añadir algún valor adicional. Por ejemplo, la supervivencia prolongada (> 5 años) se asoció con edad menor de 60 años y la supervivencia inferior a 2 años, con edad superior a 60 años. La citogenética, que tiene una influencia clave en el pronóstico, no se incluyó en el ISS, dado que hasta ese momento sólo estaba disponible en centros de referencia. Afortunadamente, en nuestro país el Grupo Español de Mieloma ha facilitado el acceso a esta información en todos los pacientes.

Tratamiento
Al abordar el tratamiento del MM, lo primero que hay que considerar es qué pacientes deben tratarse, ya que aquellos con GMSI, MM asintomático y MM sin respuesta o que no progresa pueden permanecer estables durante muchos años sin requerir tratamiento. Una vez que se considera que el paciente es sintomático, hay que decidir el tratamiento óptimo para erradicar el clon tumoral y el tratamiento de soporte. Dentro de la primera vertiente, los pacientes deben considerarse desde 2 puntos de vista: los que podrían recibir quimioterapia en dosis altas con rescate con células progenitoras (trasplante), y los que por edad o enfermedades asociadas no son candidatos a esta estrategia. Para el final dejaremos el tratamiento de los pacientes resistentes.
Tratamiento de primera línea en pacientes candidatos a trasplante autólogo
Estos pacientes son los jóvenes, que se definen en general por tener una edad inferior a 65 años, aunque este límite es objeto de debate (con buen estado general se puede ampliar a 70 años). No obstante, no hay que olvidar que no puede haber una comorbilidad que impida el futuro trasplante (insuficiencia cardíaca, respiratoria o renal, alteraciones psiquiátricas, etc.). Tras decidir la inclusión en este apartado, el tratamiento empieza a parecerse al de las leucemias agudas, ya que consta de los pasos que a continuación se comentan.
Inducción a la remisión. En pacientes jóvenes, el mejor tratamiento de inducción es aquel que consiga la mayor tasa de remisiones sin toxicidad para las células progenitoras. Esto ha hecho que el uso de alquilantes (en especial melfalán) se haya limitado al máximo en este período. Con esquemas como VAD (vincristina, doxorubicina, dexametasona)31,32, C-CAMP (carmustina, ciclofosfamida, doxorrubicina, melfolán y prednisolona)33, DT-PACE (dexametasona, talidomida, cisplatino, doxorubicina, ciclofosfamida y etopósido)34 o VBCMP/VBAD (vincristina, carmustina, ciclofosfamida, melfalán, prednisona/ vincristina, carmustina, doxorubicina, dexametasona)35, que carecen de melfalán o llevan dosis bajas, se consiguen tasas de remisión en torno al 70-80% (un 20% de respuestas completas). Con el fin de aumentar aún más la eficacia, se están ensayando esquemas basados en nuevos fármacos, tales como talidomida y dexametasona (Thal/Dex)36,37; talidomida, ciclofosfamida y dexametasona (ThaCyDex)38; lenalidomida y dexametasona (Rev/Dex)39, o bortezomib y dexametasona (Vel/Dex)40 (tabla 4)41-45. Con estos esquemas se obtienen tasas de remisión de alrededor del 90%, con respuestas completas o casi completas en el 6-38%, sin lesionar las células progenitoras, hecho que facilita la recolección de progenitores y el trasplante. No obstante, el uso de estas sustancias en fases iniciales del tratamiento debe considerarse todavía experimental, ya que también hay datos que avalan una posible utilización tardía46.

Consolidación (trasplante):
1. Trasplante con progenitores hematopoyéticos autólogos. El trasplante autólogo es hoy por hoy el tratamiento de elección del MM en pacientes que no tienen ninguna contraindicación. Esta afirmación se basa en 2 estudios en los que el trasplante autólogo con dosis altas de melfalán se tradujo en una mayor calidad de las respuestas y mejor supervivencia que la quimioterapia estándar31,33. Sin embargo, hay hasta 3 estudios, incluidos el español y el americano, en los que no se ha observado esta superioridad35,47,48. En cualquier caso, en lo que sí parece haber acuerdo es en que el trasplante mejora la respuesta de la quimioterapia inicial, generalmente duplicando el porcentaje de respuesta completa, que subiría hasta el 30-40%, y medianas de supervivencia de aproximadamente 5 años. Por ello, la clave probablemente está en el diseño de tratamientos de consolidación/mantenimiento que prolonguen la duración de las respuestas. Con respecto al régimen de acondicionamiento, el estándar es 200 mg/m2 de melfalán, ya que otros regímenes de acondicionamiento (melfalán y busulfán, melfalán e irradiación corporal total, etc.) no han mejorado la eficacia y se han mostrado más tóxicos. La utilización de doble trasplante es discutible, y los resultados del Intergroupe François du Myélome indican que sólo se benefician los pacientes que no alcanzan respuesta completa con el primero49.
La fuente de progenitores más adecuada es la sangre periférica tras estimulación con factor estimulante de colonias de granulocitos (G-CSF), con o sin ciclofosfamida. La adición de este quimioterápico no añade mucha eficacia antitumoral, pero sí mejora las cifras de la colecta de células, aunque con G-CSF sólo en dosis de 8-10 µg/kg/día generalmente se consigue suficiente celularidad50. Por último, la utilidad de procesos de selección celular tanto positiva como negativa no se ha demostrado.
2. Trasplante alogénico. La utilización de una fuente de células progenitoras sana y con capacidad de efecto de injerto contra mieloma resulta muy atractiva, en especial tras una buena remisión con un trasplante autólogo previo. Sin embargo, dadas la edad de presentación del MM y la dificultad de encontrar un donante, esta opción terapéutica está restringida a pocos pacientes (5-10%). La mortalidad del procedimiento se ha rebajado hasta un 20% o incluso menos, en especial utilizando la sangre periférica como fuente de células progenitoras y estrategias de acondicionamiento no mieloablativas51. La enfermedad del injerto contra el huésped crónica que aparece con frecuencia en estos pacientes es un inconveniente importante, pero tiene la ventaja de que los pacientes que la presentan mejoran su supervivencia al presentar efecto de injerto contra mieloma51. No obstante, el trasplante alogénico sigue siendo un tratamiento experimental y debe quedar restringido a ciertos pacientes en el contexto de ensayos clínicos.
Mantenimiento. Una vez obtenida una respuesta, y cuando el componente monoclonal ha desaparecido o permanece estable, el tratamiento combinado con quimioterapia no aporta ninguna ventaja, por lo que debe dejar paso a estrategias inmunomoduladoras, donde los nuevos fármacos tienen más posibilidades. En el metaanálisis del Myeloma Trialists' Collaborative Group52 se demostró que la utilización de interferón en este momento permite prolongar la duración de la respuesta y mejorar la supervivencia, con una media de 6 meses de ganancia, aunque a costa de una toxicidad muchas veces inaceptable. Estos resultados parecen haber sido superados con talidomida, ya que, en un estudio reciente del grupo francés en pacientes de muy mal pronóstico tratados con doble trasplante, el mantenimiento con este fármaco mejoró la supervivencia a los 4 años en un 10%, elevándola hasta un 87%, cifra muy superior a las que hasta ahora teníamos como referencia32. Una vez más, el mayor beneficio era para los pacientes que sólo habían obtenido una respuesta parcial tras los trasplantes, en los que la talidomida logró inducir una respuesta completa.
Tratamiento de primera línea en pacientes excluidos de trasplante autólogo
En este apartado incluimos a los pacientes mayores de 65 años o con situaciones de comorbilidad que impiden la realización de un trasplante de progenitores hematopoyéticos.
Inducción a la remisión. Nuevamente intentaremos utilizar un tratamiento que con la menor toxicidad induzca la mayor tasa de remisiones. La consideración de tratamiento de referencia que tenía el esquema melfalán y prednisona (MP; 9 mg/m2/día de melfalán y 60 mg/m2/día de prednisona, durante 4 días) empieza a tambalearse. Está claro que es más eficaz que otras opciones similares (p. ej., dexametasona sola) o menos tóxico (melfalán y dexametasona), y la poliquimioterapia no ha demostrado ser claramente superior. Sin embargo, la introducción de nuevos fármacos ha mejorado los resultados del esquema MP. En un estudio reciente del grupo italiano, el uso de MP y talidomida mejoró la tasa de respuesta (un 76 frente a un 54%), la supervivencia sin evento (el 54 frente al 27% a los 2 años) y, al parecer, la supervivencia global (un 80% a 3 los años) respecto a la utilización de MP solo42. El grupo francés presentó en el congreso de la Sociedad Americana de Oncología Clínica de 2006 resultados semejantes con el mismo esquema, que fue superior tanto a MP como a dosis intermedias de melfalán (100 mg/m2), seguidas de trasplante de progenitores hematopoyéticos. Utilizando talidomida y dexametasona con doxorubicina convencional43 o doxorubicina liposómica pegilada (PegLD)53 se ha llegado a describir hasta un 88% de respuestas (un 34% completas y un 24% casi completas).
El bortezomib ha sido el segundo de los nuevos fármacos que se han combinado en pacientes de nuevo diagnóstico con más de 65 años. El grupo español ha publicado un 88% de respuestas (un 32% de respuestas completas y un 11% de respuestas casi completas) con la combinación de MP y bortezomib44. Estos resultados no tienen precedentes en estos pacientes, ya que sólo se obtenían con dosis altas de quimioterapia, de las que generalmente estaban excluidos, aunque deben confirmarse en un estudio aleatorizado, que ya está en marcha.
Con lenalidomida la experiencia es menor, ya que sólo hay un estudio en pacientes de nuevo diagnóstico, que incluye tanto a candidatos como a excluidos para trasplante39. No obstante, los resultados son excelentes, ya que la asociación con dexametasona proporciona una tasa de respuestas del 91%, con un 6% de respuesta completa y un 32% de muy buena respuesta. Hay otras combinaciones prometedoras, como la asociación con MP o con PegLD53, aunque todavía quedan algunos años para conocer con exactitud la verdadera utilidad de estos fármacos en la primera línea de tratamiento.
Tratamiento de rescate
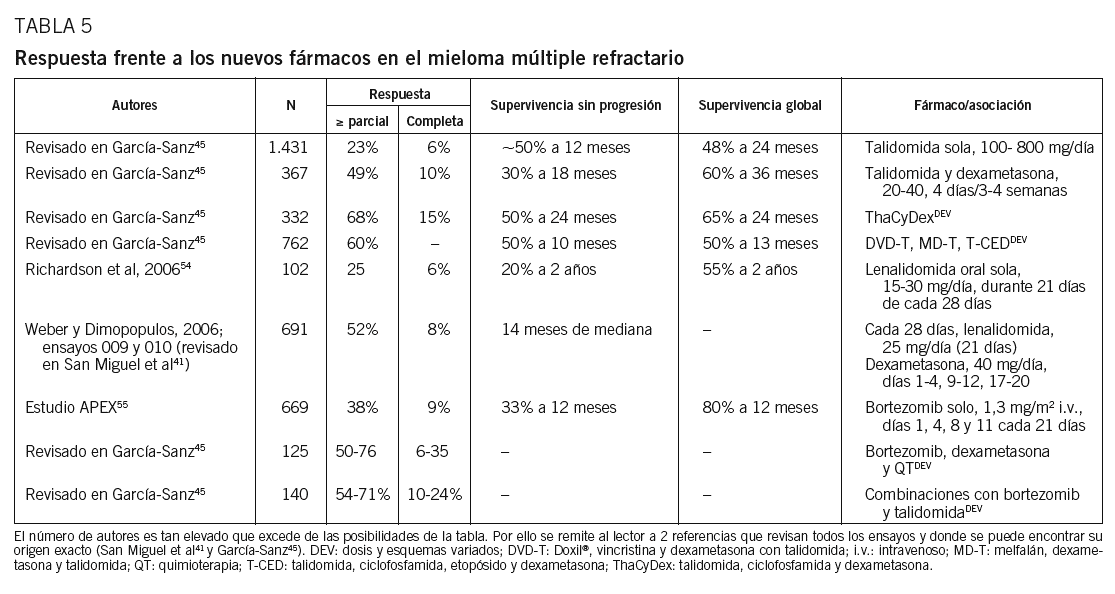
Es uno de los apartados donde más cambios ha habido. Las antiguas estrategias han quedado totalmente superadas por las combinaciones con nuevos fármacos. Así, aunque todavía se puede usar el régimen VAD y estrategias similares, las respuestas no superan el 30% y en general son cortas. En cambio, el uso de talidomida, bortezomib o lenalidomida, especialmente en combinación con alquilantes y dexametasona, permite obtener tasas de respuesta del 60-70% con toxicidad aceptable, incluso después de haber fracasado ante un trasplante simple o doble, autólogo o alogénico. Dado que la gran cantidad de bibliografía existente excede las posibilidades de esta revisión, remitimos al lector que requiera un estudio más exhaustivo a 2 recientes revisiones de nuestro grupo sobre este tipo de fármacos41,45.
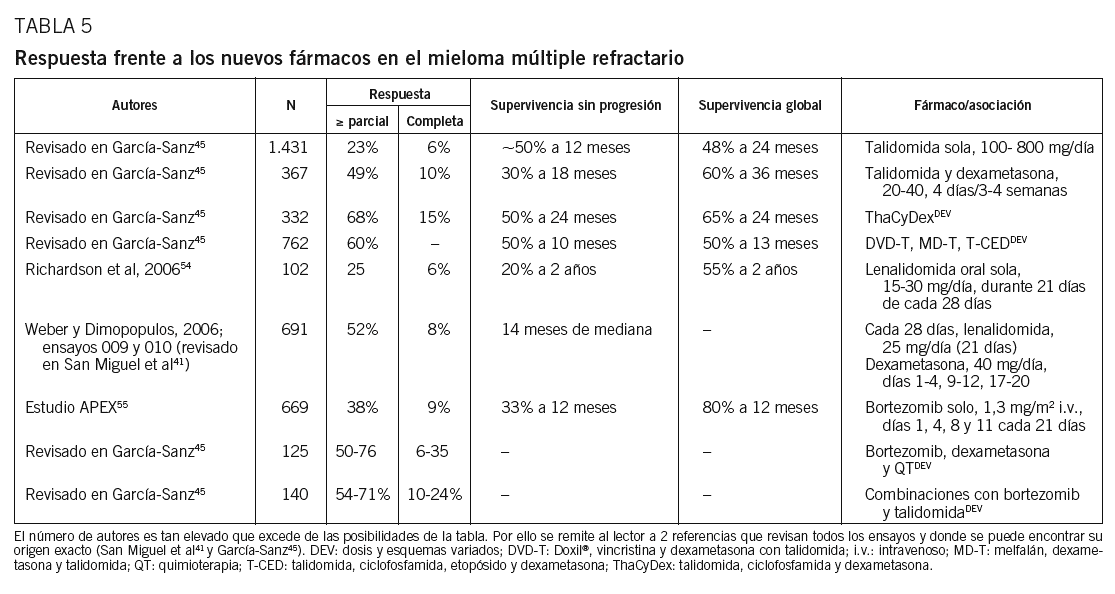
Talidomida en tratamiento de rescate. Utilizada como agente único, consigue un 30-40% de respuestas parciales, cifra que asciende hasta el 50% cuando se asocia con dexametasona (tabla 5). Sin embargo, el régimen que mejores resultados ha dado es la asociación de talidomida, ciclofosfamida y dexametasona (ThaCyDex o CTD), ya que las respuestas llegan al 65-70%, permitiendo utilizar después el trasplante como consolidación. La talidomida se utiliza por vía oral en dosis iniciales de 100 mg/día administrados por la noche. La dosis puede incrementarse hasta 400 mg/día, aunque así es más tóxica, sobre todo a largo plazo. Ello refuerza el valor de las dosis bajas o intermitentes, ya que puede ser eficaz en dosis de 50 mg/día o en cursos intermitentes de 4 días cada 2 semanas41,45.
Los efectos tóxicos más frecuentes de la talidomida son somnolencia y estreñimiento (un 30-40% de los pacientes), con una clara relación con la dosis. Sin embargo, la toxicidad más preocupante es la neuropatía periférica, distal, simétrica y de predominio motor, que puede ser irreversible y no guarda relación con la dosis, aunque es más frecuente en tratamientos prolongados.
El riesgo de trombosis venosa profunda (TVP) es un efecto secundario de la talidomida que está en el centro del debate científico. La frecuencia de este problema varía desde el 0 al 25% según las series. Es más frecuente en primera línea, si se asocia con dexametasona o quimioterapia (en especial adriamicina) y en los primeros 3-4 meses de tratamiento. Por otro lado, no hay acuerdo en cómo prevenirla. La aparición de TVP aumenta la morbilidad, pero sin aumentar la mortalidad, por lo que algunos autores recomiendan no utilizar profilaxis y únicamente tratar si aparece la complicación. Además, no hay acuerdo en qué usar como profilaxis: aspirina (81 o 325 mg/día), pese a que siempre se la ha considerado ineficaz en la profilaxis de la TVP; heparina de bajo peso molecular (equivalente a 40 mg/día de enoxaparina), o anticoagulación oral convencional (acenocumarol para mantener un cociente internacional normalizado de 2 a 3)45. En cualquier caso, la profilaxis reduce significativamente el riesgo trombótico a menos del 5%.
Bortezomib en el tratamiento de rescate. El bortezomib (Velcade® o PS-341), que se presenta como éster bórico de manitol, inhibe irreversiblemente el proteasoma 26S. Esto bloquea la degradación de proteínas ubiquitinadas, entre las que se encuentran los inhibidores del factor nuclear *B, hecho que se traduce en un efecto antiproliferativo y proapoptótico. También inhibe moléculas de adherencia, la angiogénesis, la reparación del ADN y el desdoblamiento proteico.
Los primeros ensayos clínicos con bortezomib56,57 se efectuaron en pacientes refractarios o en recaída. En ellos la tasa de respuestas fue del 35% (un 10% de respuesta completa) usando bortezomib en dosis de 1,3 mg/m2 los días 1, 4, 8 y 11 cada 21 días, pudiendo asociarse con dexametasona en caso de respuesta subóptima. Estos datos se confirmaron en un ensayo en fase III (APEX)55 con 669 pacientes con MM refractario, en quienes el bortezomib fue más eficaz que el tratamiento de rescate convencional con dexametasona en dosis elevadas, tanto en lo que se refiere a la tasa de respuesta (un 43 frente al 18%) como al tiempo hasta la progresión (6,2 frente a 3,4 meses) y la supervivencia global al año (un 80 frente al 67%). Obviamente, el paso siguiente fue combinar bortezomib con otros agentes tales como melfalán, dexametasona, adriamicina o PegLD, que permitieron alcanzar tasas de respuesta cercanas al 70-80%. También se están usando protocolos que combinan talidomida y bortezomib, pero todavía es muy pronto para extraer conclusiones definitivas41.
Entre los efectos secundarios más frecuentes figuran fatiga, síntomas gastrointestinales, trombocitopenia cíclica y, especialmente, neuropatía periférica. Ésta suele ser distal, simétrica y sensitiva, en forma de dolor neuropático. Es importante conocer bien los efectos secundarios para detectarlos e instaurar de forma precoz las reducciones de dosis o el tratamiento necesarios.
Tratamiento con lenalidomida en el mieloma múltiple refractario. La lenalidomida es un derivado de la talidomida con la ventaja a priori de que es más potente in vitro y menos tóxico. Como fármaco único conseguía un 25% de respuestas en pacientes de muy mal pronóstico (recaídas de trasplante, tercera línea o posterior, etc.)54. Sin embargo, los resultados han sido mejores en ensayos en fase III, asociada a dexametasona, con respuestas superiores al rescate convencional (un 60 frente a un 22%) y prolongación significativa de la supervivencia sin progresión (14 frente a 5 meses) y global (29 frente a 20 meses). La toxicidad de la lenalidomida parece menor que la de talidomida (en cuanto a sedación, estreñimiento y neuropatía), pero también provoca TVP y neutropenia de grado moderado a grave39,41,54.
Evaluación de la respuesta
Hasta hace poco los criterios de respuesta más usados eran los del European Blood and Marrow Transplant (EBMTR), generalmente conocidos como criterios de Bladé, publicados en 1998. Sin embargo, ahora hay una mejor calidad en las respuestas y es posible efectuar el seguimiento de los pacientes con métodos nuevos (citometría de flujo, reacción en cadena de la polimerasa, FLC, etc.), lo que ha impulsado al grupo internacional de mieloma a revisar estos criterios58. En especial, se han creado una nueva categoría de respuesta completa, denominada «respuesta completa estricta», y la categoría «muy buena respuesta parcial o respuesta casi completa», y se ha eliminado la respuesta menor, equiparándola con la enfermedad estable. En la tabla 6 se muestra la definición completa de estos nuevos criterios.

Tratamiento de soporte
Tan importante como el tratamiento de la enfermedad es el control de los síntomas y complicaciones del MM, que también ha cambiado en los últimos años. De hecho, parte de la mejoría en la supervivencia de los pacientes debe atribuirse a la reducción de la mortalidad derivada de infecciones, hipercalcemia e IR. Además, este aspecto del tratamiento permite mejorar la calidad de vida, cada vez más valorada por los pacientes.
Tratamiento del dolor óseo. Para reducir el dolor óseo lo más eficaz es controlar la enfermedad de base, pero es frecuente tener que recurrir a los analgésicos, desde el paracetamol a la morfina, asociados o no a antiinflamatorios y relajantes musculares. Dados los problemas renales de estos pacientes, se prefiere evitar los antiinflamatorios no esteroideos. Si el dolor es debido a una lesión localizada, puede emplearse radioterapia (2.000-3.000 cGy); si es por aplastamiento vertebral, están indicadas la vertebroplastia o la cifoplastia, sin olvidar los accesorios ortopédicos7. Los bisfosfonatos pueden ser útiles, pero su efecto debe considerarse a largo plazo7.
Fracturas óseas y compresión medular. Lo mejor es prevenirlas mediante ejercicio físico regular y bisfosfonatos (4 mg de ácido zoledrónico o 90 mg de pamidronato por vía intravenosa cada mes), que disminuyen las complicaciones óseas y retrasan su aparición. Estos fármacos son muy útiles, pero tienen limitaciones, sobre todo porque favorecen el desarrollo de osteonecrosis de mandíbula en tratamientos a largo plazo, por lo que su uso debería restringirse a un máximo de 2 años y esperar a reintroducirlos cuando reaparezca la enfermedad activa. Las lesiones de huesos largos son subsidiarias de fijación ortopédica y, en ocasiones, quirúrgica. La compresión medular es una urgencia que se trata con dexametasona y radioterapia local; si es el síntoma de presentación y todavía no hay diagnóstico de MM, debe realizarse cirugía descompresiva y estudio anatomopatológico7.
Hipercalcemia. Es otra urgencia en el mieloma, ya que favorece el desarrollo de IR. Siempre hay que garantizar una buena hidratación (2-4 ml/kg/h de suero fisiológico) y son beneficiosos los diuréticos de asa y los esteroides, pero el tratamiento actual más eficaz (respuesta en más del 90%) es el ácido zoledrónico en dosis única de 4 mg en infusión intravenosa de 15 min2.
Insuficiencia renal. Hay que prevenirla con hidratación, alcalinización y administración de quimioterapia, corrigiendo la hipercalcemia y las infecciones y evitando los nefrotóxicos. Si el paciente se presenta con IR aguda, hay que adoptar las medidas habituales y evitar protocolos con melfalán, que obligan a ajustar la dosis (usar VAD, VBAD, o los nuevos fármacos como bortezomib) y, si es necesario, indicar hemodiálisis o diálisis peritoneal. Si la IR es grave e irreversible y el mieloma responde al tratamiento, se debe recurrir a la diálisis crónica sin desestimar la posibilidad de un trasplante renal7.
Anemia. Se debe corregir de forma aguda (transfusión) cuando sea claramente sintomática y el valor de hemoglobina menor de 8 g/dl. La utilización de agentes estimulantes del receptor eritropoyético debe ensayarse cuando la hemoglobina sea inferior a 10-11 g/dl, pero siempre hay que descartar causas concretas de anemia (déficit de hierro, ácido fólico, B12, hemorragias, etc.). Lo más conveniente sería usar dosis semanales (NeoRecormon®: 30.000 UI; Eprex® o Epopen®: 40.000 UI; AraNESP®: 150 µg), bisemanales (AraNESP®: 300 µg) o trisemanales (AraNESP® 500 µg) por vía subcutánea, aunque hay otros esquemas. Si la respuesta no es adecuada, se puede doblar las dosis antes de dar por fracasado el tratamiento. En cualquier caso, se debe seguir las recomendaciones sobre el uso de agentes eritropoyéticos en este tipo de pacientes que se establecen en las guías, como las que se revisan en la referencia número 5.
Infecciones. Con el tratamiento habitual la neutropenia grave es poco frecuente, por lo que el uso de G-CSF sólo se justifica en casos seleccionados. Cuando se presenta una infección, hay que emplearvulánico, piperacilina-tazobactam, etc.), evitando los nefrotóxicos como los aminoglucósidos. En caso de infecciones recurrentes, puede considerarse la administración profiláctica de gammaglobulina si hay hipogammaglobulinemia policlonal grave7, aunque no es una práctica habitual.
Conclusiones
Aunque el concepto fundamental del MM no ha cambiado desde las primeras descripciones de la enfermedad, en la actualidad hay que revisar los siguientes conceptos:
El diagnóstico del mieloma se basa en la presencia de componente monoclonal e infiltración plasmocitaria tisular, independientemente de su cuantía, pero asociada a lesiones orgánicas, especialmente en forma de hipercalcemia, IR, anemia o lesiones óseas.
La fisiopatogenia de la enfermedad se encuentra asociada a lesiones genéticas en células de línea linfoide B que provocan incrementos de la cantidad de ADN (hiperploidía) y/o translocaciones cromosómicas en la región de cambio de isotipo del gen de la cadena pesada de las Ig. Esto desencadena al final un fenómeno oncogénico común: sobreexpresión de una ciclina D (D1, D2 o D3).
El tratamiento del mieloma se basa en la inducción, que de momento incluye quimioterapia convencional, seguida de trasplante autólogo con melfalán en dosis altas, si el paciente puede tolerarlo. No obstante, junto a la quimioterapia convencional hay que considerar nuevos fármacos (talidomida, bortezomib o lenalidomida). En pacientes que no puedan recibir un trasplante, la inducción debe basarse en esquemas clásicos como MP o dosis altas de dexametasona, probablemente asociados a alguno de los nuevos fármacos como la talidomida o el bortezomib. El tratamiento de rescate debe incluir necesariamente alguno de los 3 nuevos fármacos, idealmente combinado con otros. El trasplante alogénico sigue siendo, hoy por hoy, una estrategia experimental.
El tratamiento de soporte de los pacientes con mieloma debe contemplar el uso de bisfosfonatos para tratar la hipercalcemia y prevenir las complicaciones esqueléticas, el empleo de eritropoyetina en los pacientes anémicos, la utilización de cifoplastia o vertebroplastia en lesiones vertebrales graves y el tratamiento antibiótico enérgico en caso de infecciones.
