




La paciente era alérgica a la penicilina y las sulfamidas, fumaba 10 cigarrillos diarios y no consumía alcohol. Se le habían realizado una salpingectomía y drenaje de un absceso tubárico como consecuencia de una enfermedad inflamatoria pélvica 4 años atrás, con 2 ingresos posteriores, hacía 3 y 2 años, por recidiva de la infección, que en ambos casos se resolvió con tratamiento antibiótico. Había iniciado tratamiento de fertilización in vitro un año atrás, con transferencia de embriones en 4 ocasiones, aunque sólo se implantaron en 2 y ambos concluyeron en sendos abortos espontáneos que no requirieron legrado. Se realizó una determinación de anticuerpos antifosfolipídicos y anticoagulante lúpico, que fue negativa, además de un estudio de hemostasia, que fue normal. Por último, también presentaba anemia ferropénica detectada hacía un año, con valores de hematocrito de hasta 0,30l/l. Se la había estudiado en otro centro mediante fibrogastroscopia y biopsias duodenales, en las que se encontró un infiltrado linfocitario con un 60% de linfocitos CD8+, que se consideró indicativo de celiaquía. Se le recomendó una dieta sin gluten, pero no la realizó ni siguió más controles médicos.
Al cabo de un año la paciente ingresó en otro centro para estudio por presentar fiebre diaria de hasta 38,8°C, sin foco aparente, de 2 meses de evolución, que se había iniciado tras el último aborto. No refería leucorrea. También presentaba discretas molestias abdominales que se localizaban en mesogastrio e hipogastrio, pero sin náuseas, vómitos ni diarrea. Refería astenia, anorexia y pérdida ponderal de 15kg de peso. De las pruebas realizadas durante aquel ingreso destacaban los siguientes datos: velocidad de sedimentación globular (VSG) de 123mm/h, hemoglobina de 67g/l, volumen corpuscular medio de 63fl, leucocitos de 7,6×109/l, plaquetas de 762×109/l, sideremia de 4μg/dl y ferritina de 32ng/ml. Las serologías para citomegalovirus, virus de Epstein-Barr, Toxoplasma y Brucella, 2 hemocultivos y los anticuerpos anti-ADN y antitransglutaminasa fueron negativos. En una ecografía ginecológica se observó una pequeña cantidad de líquido en el fondo del saco de Douglas, sin otras alteraciones. Se realizó una fibrogastroscopia, que fue normal, y las biopsias efectuadas evidenciaron gastritis crónica sin presencia de Helicobacter pylori. Una tomografía computarizada (TC) abdominal puso de manifiesto, además del mencionado líquido en el fondo del saco de Douglas, 3 imágenes hepáticas de 1–2cm de diámetro que se orientaron como hemangiomas. La exploración ginecológica fue normal. Se inició tratamiento antibiótico con metronidazol (500mg/8h) y ceftriaxona (1g/24h). La paciente continuaba con febrícula 10 días después del ingreso. En ese momento solicitó el alta voluntaria y el mismo día acudió a urgencias del Hospital Clínic de Barcelona, desde donde ingresó para estudio.
En la exploración física destacaban palidez y temperatura de 37,9°C. El examen ginecológico fue normal y en la ecografía transvaginal se observó una moderada cantidad de líquido libre en el fondo del saco de Douglas, así como una imagen paraanexial derecha heterogénea de 39×26mm. La paciente no accedió a que se le practicara una culdocentesis. La exploración por aparatos fue normal. En los análisis de sangre destacaban: VSG de 144mm/h, proteína C reactiva de 15mg/dl, hematíes de 3,78×1012/l, hemoglobina de 70g/l, hematocrito de 0,25l/l, volumen corpuscular medio de 65fl, hemoglobina corpuscular media de 18pg, leucocitos de 12,3×109/l (60% segmentados, 3% no segmentados, un 3% eosinófilos, un 1% basófilos, un 27% linfocitos y un 6% monocitos), plaquetas de 881×109/l, actividad de protrombina del 73%, tiempo de cefalina de 36s, glucemia de 80mg/dl (4,43mmol/l), nitrógeno ureico en sangre de 8mg/dl (1,3mmol/l), creatinina de 0,7mg/dl (62μmol/l), sodio de 135mmol/l, potasio de 4,4mmol/l, calcio de 9,4mg/dl (2,3mmol/l), fósforo de 3,8mg/dl (1,3mmol/l), sideremia de 6μg/dl (1,1μmol/l), aspartato-aminotransferasa de 14U/l, alanina-aminotransferasa de 4U/l, lactatodeshidrogenasa de 327U/l (valor normal [VN]: 250–450), gammaglutamil transpeptidasa de 174U/l, fosfatasa alcalina de 712U/l, bilirrubina total de 0,8mg/dl (13,6μmol/l), amilasa de 37U/l, lipasa de 36U/l, colesterol de 120mg/dl (1,4mmol/l), triglicéridos de 94mg/dl (1,0mmol/l), creatincinasa de 22U/l y proteínas totales de 70g/l (albúmina: 47%; alfa-1-globulinas: 6%; alfa-2-globulinas: 17%; betaglobulinas: 11%; gammaglobulinas: 19%). El sedimento de orina, el electrocardiograma, la radiografía de tórax y la de abdomen fueron normales. La detección de sangre oculta en heces resultó positiva. Se inició tratamiento con clindamicina (600mg/8h), gentamicina (240mg/24h), hierro intravenoso y dieta sin gluten.
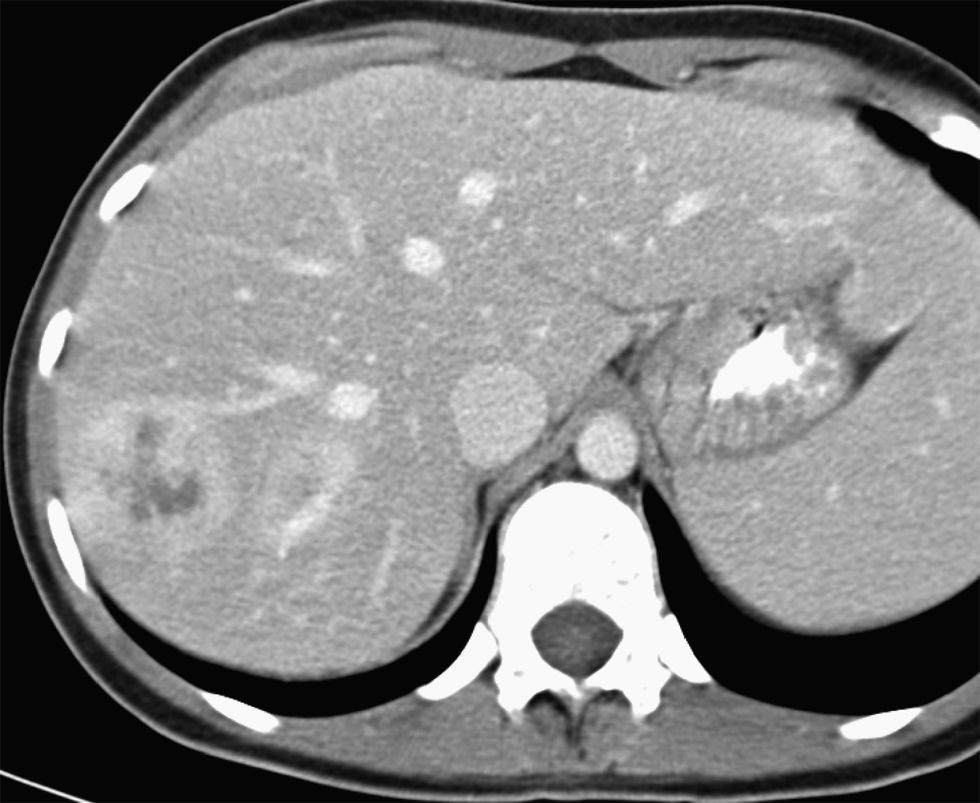
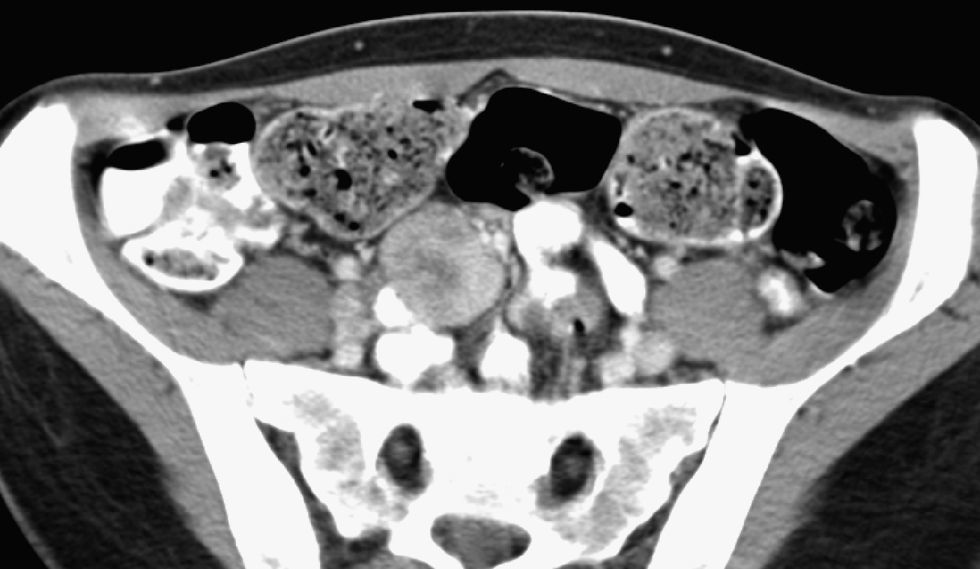
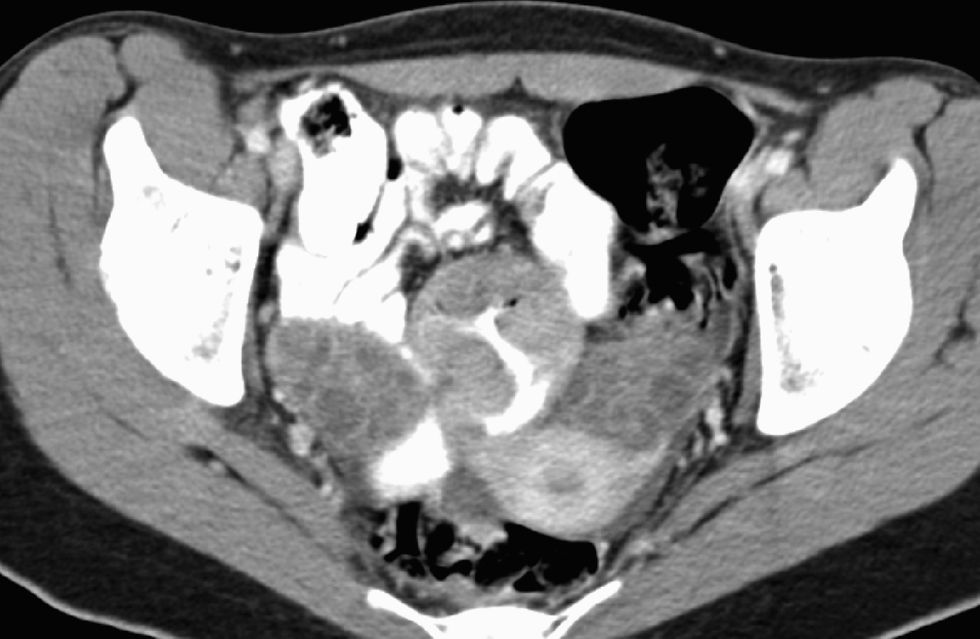
Durante los días siguientes se le practicaron una fibrocolonoscopia, que fue normal, y una fibrogastroscopia, en la que destacaba atrofia de las vellosidades duodenales. La biopsia duodenal evidenció un patrón vellositario preservado, discreto infiltrado linfoplasmocitario en la lámina propia y algunos leucocitos intraepiteliales. La transferrina fue de 1,19g/l (VN: 1,9–3,1); el receptor soluble de la transferrina, de 6,8mg/l (VN: 0,8–1,8); la capacidad total para ligar hierro, de 1,7mg/l (VN: 2,5–4,5), y la saturación de la transferrina, del 6%. En una ecografía abdominal se observaron 2 áreas hipoecogénicas en el hígado, de 3,5 y 1,5cm, de difícil delimitación, y escasa cantidad de líquido libre intraabdominal; el resto de la exploración fue normal. En la TC torácica no se observaron hallazgos relevantes. En la TC abdominopélvica se evidenciaron 5 lesiones focales hepáticas de aspecto heterogéneo, con captación periférica y centro hipodenso (fig. 1). En la pelvis se observaron 3 masas nodulares heterogéneas que se localizaban en la raíz del mesenterio (fig. 2), de 3,5cm de diámetro máximo, y un acusado engrosamiento de un tramo del íleon que afectaba unos 4cm de longitud, sin condicionar estenosis de la luz ni dilatación de las asas proximales (fig. 3). Los ovarios estaban aumentados de tamaño, con múltiples quistes de pequeño tamaño, y había una mínima cantidad de líquido en la pelvis menor. Los hemocultivos (×4), el urinocultivo y el coprocultivo fueron negativos. Las serologías frente a los virus de la hepatitis B y C, de la inmunodeficiencia humana, lúes, Mycoplasma pneumoniae, Rickettsia conorii, Coxiella burnetii y Borrelia burgdorferi fueron negativas. Los títulos de inmunoglobulinas fueron normales, al igual que los de hormonas tiroideas. Los anticuerpos antinucleares, cuyo valor era de 80 URF (formas recombinantes únicas), presentaban un patrón homogéneo y parcheado; los valores de antimúsculo liso fueron de 1/80, los anti-ADN bicatenario de 1U/ml y los antimitocondriales, anticélula parietal gástrica, antimicrosómicos contra hígado y riñón, anticitoplásmicos de neutrófilo, antimieloperoxidasa, antiproteinasa-3, antirreticulina, antigliadina, antitransglutaminasa y antiendomisio, así como el factor reumatoide, negativos. El test de la xilosa y el de la elastasa fueron normales. Se determinaron la beta-2-microglobulina, la alfafetoproteína, el antígeno carcinoembrionario, CA 153, CA 125, CA 19.9, CYFRA 21/1, la enolasa neuronal específica y el TAG 72, que fueron normales.



La paciente, después de 25 días de ingreso, permanecía con dolor abdominal discreto, localizado en el mesogastrio, que no guardaba relación con la ingesta. A pesar de haber sustituido los antibióticos iniciales por doxiciclina a dosis de 100mg/12h, continuaba con fiebre intermitente de hasta 39°C, bien tolerada, sin escalofríos, y con hemocultivos repetidamente negativos. La anemia había empeorado hasta alcanzar un valor de hematocrito de 0,21l/l, por lo que se transfundieron 2 unidades de concentrados de hematíes.
A continuación se realizó una prueba que resultó diagnóstica.
Diagnóstico DiferencialDr. Xavier Bessa. En resumen, se trata de una paciente de 21 años, con antecedentes de abortos espontáneos de repetición tras episodios de fertilización in vitro, a quien hacía aproximadamente un año se le había detectado una anemia ferropénica atribuida a una probable celiaquía sobre la base del hallazgo de un porcentaje de linfocitos CD8 del 60% en las biopsias duodenales. Se le aconsejó una dieta exenta de gluten, que la paciente no realizó. La consulta médica se efectuó un año más tarde, cuando la paciente presentó clínica de síndrome febril persistente, de 2 meses de evolución, junto con una acusada pérdida ponderal. En la exploración física no se apreciaron linfadenopatías periféricas y en los análisis se detectó anemia microcítica con reactantes de fase aguda (trombocitosis, elevación de la VSG y la proteína C reactiva) y colestasis disociada (elevación de la gammaglutamil transpeptidasa y fosfatasa alcalina con bilirrubina normal). Entre las pruebas de imagen efectuadas —TC y ecografía abdominal—, destaca la existencia de al menos 5 lesiones focales hepáticas de aspecto heterogéneo, con captación periférica y centro hipodenso (una TC abdominal previa había mostrado únicamente 3 imágenes de 1–2cm, orientadas como hemangiomas), así como 3 masas nodulares heterogéneas de 3,5cm de diámetro que se localizaban en la raíz del mesenterio, y un marcado engrosamiento de un tramo del íleon que afectaba unos 4cm de longitud. Antes de proseguir, agradecería a la radióloga que comentara la iconografía del caso.
Dra. Carmen de Juan. La radiografía de tórax, la de abdomen y una TC torácica no mostraron hallazgos patológicos. En una ecografía se observaron 3 lesiones focales hepáticas que eran hiperecogénicas y mal definidas, de 4cm de diámetro máximo, inespecíficas. La TC abdominopélvica evidenció 5 lesiones focales hepáticas de aspecto heterogéneo y de comportamiento hipervascular, con periferia hipercaptante y un centro hipodenso de aspecto necrótico, de 4cm de diámetro máximo. En la fase retardada el área hipercaptante de estas lesiones focales hepáticas se hacía prácticamente isodensa con el parénquima, pero el centro permanecía hipodenso. En la pelvis menor, en la raíz del mesenterio, se observaron 3 masas sólidas heterogéneas e hipercaptantes, de 3,5cm de diámetro máximo, que presentaban un comportamiento radiológico similar al de las lesiones focales hepáticas. Adyacente a estas masas mesentéricas se observaba un segmento de un asa ileal de unos 4cm de longitud, que mostraba unas paredes notablemente engrosadas y que englobaba la totalidad de la circunferencia del segmento afectado. La lesión no condicionaba estenosis de la luz intestinal ni dilatación de las asas proximales. Se observaba también mínima cantidad de líquido libre en la pelvis menor.
Dr. X. Bessa. Por lo tanto, a la hora de establecer el diagnóstico diferencial nos basaremos en las entidades que pueden cursar con una clínica similar y que ocasionan un engrosamiento segmentario y nodular del íleon, adenopatías mesentéricas y lesiones focales hepáticas. Globalmente, cabe considerar 3 grandes posibilidades: la inflamatoria, la infecciosa y la tumoral. No obstante, la clínica y, sobre todo, las pruebas de imagen, que muestran adenopatías mesentéricas de gran tamaño con hipodensidad central y el crecimiento, tanto en número como en tamaño, de las lesiones focales hepáticas, apuntan claramente al origen tumoral del proceso de base actual.
La entidad inflamatoria clásica que puede afectar la zona ileal es la enfermedad de Crohn1. No obstante, en ésta las manifestaciones clínicas habituales son la diarrea, frecuentemente con productos patológicos (sangre, moco o pus), y el dolor abdominal. Con frecuencia hay fiebre —en relación con la propia enfermedad o como complicación de ella, como, por ejemplo, el desarrollo de abscesos— y pérdida de peso, pero en relación con los déficit nutricionales. Esto conlleva —aparte de la elevación de los reactantes de fase aguda— hipoproteinemia e hipoalbuminemia. La afectación ileal exclusiva acontece en el 30% de los casos, pero se localiza fundamentalmente en el extremo distal del íleon, con o sin participación del polo cecal en continuidad. En las fases evolucionadas pueden aparecer linfadenopatías mesentéricas, pero de pequeño tamaño, entre 3 y 8mm. El desarrollo de adenopatías de tamaño superior a 1cm obliga a descartar un carcinoma o linfoma asociado2. Por lo que se refiere a las lesiones focales, en la enfermedad de Crohn pueden desarrollarse, tras un largo período de evolución, un adenocarcinoma y potencialmente metástasis, pero no parece ser el caso. Morfológicamente las imágenes tampoco son indicativas de abscesos y la falta de respuesta a los múltiples tratamientos antibióticos va en contra de esta opción.
Entre las causas infecciosas cabe destacar fundamentalmente 2 posibilidades: la tuberculosis intestinal y la infección por Yersinia. La tuberculosis intestinal3, no obstante, engloba de forma habitual la zona ileocecal y se manifiesta en forma de múltiples ulceraciones (60%), masa seudotumoral (10%) o mixta (30%). La clínica es larvada y habitualmente crónica en forma de dolor abdominal crónico en el 80–90% de los casos, asociado a anorexia, fatiga, fiebre, pérdida de peso, diarrea y hemorragia intestinal. En los análisis se constata elevación de la VSG en el 50–80% de los casos y anemia moderada. Los hallazgos radiológicos típicos en la TC son el engrosamiento de la zona ileocecal y una linfadenopatía mesentérica característica, con hipodensidad central en relación con la licuefacción caseosa central. La afectación hepática es excepcional y se manifiesta en forma de fiebre de origen desconocido, anorexia, pérdida de peso y síntomas abdominales inespecíficos. El desarrollo de abscesos hepáticos tuberculosos es poco frecuente. La hepatomegalia es común y los análisis evidencian hipoproteinemia y elevación de la fosfatasa alcalina y la bilirrubina4. Para establecer el diagnóstico es preciso obtener muestras histológicas de las zonas afectadas y confirmar la presencia de granulomas caseificantes. Por otro lado, Yersinia enterocolitica y Yersinia pseudotuberculosis son unos patógenos intestinales importantes que causan un espectro de enfermedades clínicas que van desde la gastroenteritis simple hasta la ileítis y colitis invasiva5. En adultos jóvenes se han descrito adenitis mesentéricas e ileítis asociadas a infección por Yersinia. La bacteriemia por Yersinia es relativamente infrecuente y se observa en pacientes con enfermedad subyacente como procesos malignos, diabetes mellitus, anemia y enfermedad hepática. En huesos, articulaciones y pulmones pueden asentarse focos metastásicos. Habitualmente los cultivos en heces permiten establecer el diagnóstico y, a pesar de que Yersinia es resistente a la penicilina y las cefalosporinas, tiene una gran sensibilidad a diferentes tratamientos como el cloranfenicol, las tetraciclinas, trimetoprima-sulfametoxazol y gentamicina, antibiótico este último que la paciente había recibido durante el ingreso.
Finalmente, centraremos la discusión en las enfermedades neoplásicas, diagnóstico más probable en esta paciente. Las neoplasias del intestino delgado son poco frecuentes y representan el 1,1–2,4% de las neoplasias gastrointestinales malignas. Las neoplasias malignas más frecuentes en el intestino delgado son los adenocarcinomas, los tumores neuroendocrinos, los linfomas y los tumores mesenquimales del tracto gastrointestinal (sarcomas o tumores de la estroma gastrointestinal [GIST])6.
Los linfomas gastrointestinales primarios representan el 5–10% de los tumores gastrointestinales7. Entre un 20 y un 30% de los linfomas, o en torno a un 5% de los linfomas no hodgkinianos periféricos en general, se localizan primariamente en el intestino delgado y se diferencian de los linfomas gástricos por las manifestaciones clínicas, el tratamiento y el pronóstico8. Estudios retrospectivos han observado que alrededor de un 60–80% de éstos son linfomas B, con frecuencia linfomas difusos grandes de células B del intestino delgado, predominantemente de la zona ileocecal9. Los linfomas de células marginales, del manto o de localización colónica, son infrecuentes10. Por el contrario, los linfomas intestinales de células T son a menudo multifocales y se localizan mayoritariamente en el yeyuno o íleon proximal11. En el pasado, la clasificación europea-americana de linfomas revisada (REAL) diferenciaba los linfomas de células T entre aquellos asociados a enteropatía (EATCL) y los no asociados a enteropatía dependiendo de la existencia de ésta en la mucosa distante al tumor12. No obstante, la nueva clasificación de los linfomas de la Organización Mundial de la Salud no mantiene esta subclasificación específica de los no EATCL, que se consideran linfomas T periféricos13.
A la hora de iniciar el diagnóstico diferencial de la paciente debe tenerse en cuenta un antecedente relevante: la anemia ferropénica. La causa más frecuente de ésta en mujeres jóvenes son las pérdidas menstruales, pero en este caso no se menciona y, por el contrario, se orientó dicha anemia como secundaria a celiaquía sobre la base del incremento de los linfocitos intraepiteliales en el duodeno. La forma de presentación de la celiaquía varía mucho según el grupo de edad afectado. En los pacientes jóvenes la enfermedad se manifiesta a menudo a través de síntomas extraintestinales como la baja estatura, los síntomas neurológicos o bien, como en el caso que nos ocupa, en forma de infertilidad o anemia. El diagnóstico de enfermedad celíaca requiere la obtención de biopsias duodenales con los hallazgos típicos de linfocitosis intraepitelial, la hiperplasia de criptas, la atrofia vellositaria y una respuesta positiva a la dieta sin gluten14. Clásicamente la enfermedad celíaca se consideraba poco prevalente, pero con el desarrollo de anticuerpos específicos contra la enfermedad se ha observado que su prevalencia es mayor de lo sospechado. Estudios epidemiológicos que utilizan estos tests con posterior verificación histológica han establecido una prevalencia de 1:300 a 1:500 en la mayoría de países15. Asimismo, su incidencia en pacientes con anemia ferropénica se ha establecido en el 2,3–5%16. La gradación de la inflamación y de los cambios en la arquitectura de la mucosa duodenal son variables en la enfermedad celíaca, y esta progresión en el daño de la arquitectura fue inicialmente descrita por Marsh et al. La gradación histológica incluye un patrón infiltrativo (Marsh I), que se caracteriza por un incremento de linfocitos intraepiteliales; un patrón hiperplásico (Marsh II), con criptas alargadas que presentan un incremento de la división celular; un patrón con marcada atrofia vellositaria (Marsh III), y un patrón hipoplásico (Marsh IV)17. No obstante, el incremento de linfocitos intraepiteliales sin otros hallazgos histológicos (Marsh I) no es específico de la enfermedad celíaca18. El incremento de los linfocitos intraepiteliales no es ni exclusivo ni patognomónico de la enfermedad celíaca19. Entre sus causas destacan la sensibilidad a otras proteínas alimentarias, las infecciones, particularmente por Giardia lamblia, Cryptosporidium, la infección por H. pylori y el sobrecrecimiento bacteriano. En la paciente de nuestro caso se ha descartado razonablemente la infestación parasitaria (hemocultivos, coprocultivos y parásitos negativos) y se ha excluido en el estudio histológico la presencia de H. pylori. Asimismo, se han efectuado múltiples pautas de tratamiento antibiótico, sin aparente respuesta. Otra de las causas de linfocitosis intraepitelial es el consumo de fármacos, especialmente los antiinflamatorios no esteroideos, que parece excluirse mediante la historia clínica. Por otro lado, varias anomalías asociadas a la alteración de los mecanismos de regulación inmunitaria se acompañan de un aumento de los linfocitos intraepiteliales (tiroiditis de Hashimoto, enfermedad de Graves, artritis reumatoide, psoriasis, esclerosis múltiple, lupus eritematoso sistémico y enteropatía autoinmunitaria). Finalmente, otras enfermedades asociadas son la hipogammaglobulinemia (deficiencia de inmunoglobulina A o inmunodeficiencia común variable) y la enfermedad inflamatoria intestinal (colitis ulcerosa y enfermedad de Crohn). De forma global, sobre la base de la historia clínica y las pruebas efectuadas (serologías, marcadores de autoinmunidad, estudios endoscópicos y otras) puede descartarse la mayoría de estas causas de linfocitosis intraepitelial.
En relación con el papel de los anticuerpos para el diagnóstico de la celiaquía, negativos en esta paciente, múltiples estudios han demostrado que la sensibilidad y especificidad de dichos anticuerpos (antitransglutaminasa tisular, antiendomisio y antigliadina) guardan relación con el grado de lesión histológica. La sensibilidad de los anticuerpos antiendomisio y antitransglutaminasa tisular del tipo inmunoglobulina A puede ser de menos del 50% en pacientes con una lesión histológica del tipo Marsh II20 y nula en pacientes con linfocitosis intraepitelial. En consecuencia, al valorar la historia de la paciente debería tenerse en cuenta la posibilidad de que nos hallemos ante una enfermedad celíaca atípica (con pocas manifestaciones gastrointestinales pero con otros problemas médicos como anemia ferropénica, osteoporosis, corta estatura o infertilidad) o latente (mucosa intestinal normal o sólo con incremento de linfocitos intraepiteliales).
Volviendo al diagnóstico previo de celiaquía en el caso que nos ocupa, es importante identificar a los pacientes celíacos que presentan mínimos cambios histológicos porque pueden desarrollar deficiencias nutricionales y además presentan un pequeño riesgo de desarrollar tumores malignos. Basándonos en los múltiples estudios epidemiológicos podemos concluir que en los pacientes celíacos no hay un incremento del riesgo de neoplasia respecto a la población general, pero determinados tumores malignos, en particular el linfoma no hodgkiniano y los carcinomas gastrointestinales, pueden estar ligeramente aumentados21, sobre todo en los pacientes que no siguen a rajatabla una dieta sin gluten. Con frecuencia los tumores malignos se diagnostican entre el primer y el tercer año del diagnóstico de la enfermedad celíaca, ya que la enfermedad neoplásica precipita el diagnóstico de la celiaquía o, por el contrario, las investigaciones llevadas a cabo para llegar al diagnóstico de la enfermedad celíaca descubren la existencia de una neoplasia oculta. Este hecho es particularmente común en el caso de una entidad infrecuente: el linfoma gastrointestinal.
La asociación entre enfermedad celíaca y linfoma no hodgkiniano intestinal de células T, llamado comúnmente EATCL, es particularmente estrecha a pesar de la rareza de estos linfomas22. Los EATCL representan menos del 1% de los linfomas no hodgkinianos. La mayoría de los estudios publicados estiman un riesgo incrementado de EATCL en los pacientes celíacos respecto a la población general de 2,7 a 6,3 veces, aunque algunos estudios han evidenciado un riesgo superior a 42 veces respecto a la población general. Múltiples estudios señalan que hay una reducción del riesgo de linfoma con una estricta observancia de la dieta sin gluten; no obstante, el beneficio de ésta comienza a acumularse de forma lenta en los pacientes diagnosticados en edades avanzadas. Debe tenerse en cuenta también que el riesgo de desarrollar un linfoma de células B intestinal (riesgo: 2,2) o un linfoma T extraintestinal (riesgo: 3,6) está aumentado en pacientes con enfermedad celíaca, aunque el riesgo es menor respecto al del clásico EATCL23. Éste afecta de forma más frecuente al varón (2:1) y la edad media en el momento del diagnóstico es de 50 años. Los síntomas más comunes en el momento de la presentación son dolor abdominal (84%), pérdida de peso (81%), diarrea (39%) o vómitos (29%). Hasta un tercio de los pacientes puede presentar fiebre y sudores nocturnos. Es relativamente frecuente su manifestación en forma de perforación intestinal u obstrucción. La afectación principal es yeyunal, aunque puede afectar a cualquier tramo del intestino delgado. De forma habitual la enfermedad se detecta en múltiples segmentos del intestino delgado o se ha diseminado a los ganglios mesentéricos, el hígado, el bazo o la médula ósea. Se han descrito linfadenopatías mesentéricas con cavitación central tanto en pacientes celíacos con EATCL como sin linfoma asociado24. Hay que destacar, además, que durante el ingreso de nuestra paciente se efectuó una nueva endoscopia digestiva, en la que se observó macroscópicamente una mucosa de aspecto atrófico, pero el estudio histológico no confirmó este dato. A pesar de que la enfermedad celíaca afecta de forma parcheada a la mucosa intestinal, si bien pudo haberse producido un error de muestreo, la negatividad de los anticuerpos contra la celiaquía no apoya el diagnóstico de ésta y, en consecuencia, de EATCL. La determinación de los antígenos de histocompatibilidad HLA DQ2 y DQ8, por su elevado valor predictivo negativo, podría haber ayudado a descartar definitivamente esta posibilidad.
El íleon distal es el lugar de afectación típico del linfoma B del intestino delgado por la elevada cantidad de tejido linfoide en esta porción del intestino delgado25. Este tipo de linfoma suele presentarse como una voluminosa masa circunferencial en la pared intestinal y habitualmente se acompaña de extensión mesentérica y a los ganglios linfáticos regionales. Un reciente estudio prospectivo que comparaba las manifestaciones clínicas de los diferentes subtipos de linfomas no ha demostrado diferencias significativas en la edad y el sexo entre los diferentes subtipos de linfoma. La fiebre y la diarrea se observaron en un tercio de los linfomas T y rara vez en los linfomas B. Por último, los linfomas T se localizaron preferentemente en el duodeno y el yeyuno, y los linfomas B se localizaron sobre todo en la zona ileal26. En relación con los hallazgos analíticos, estos pacientes suelen presentar anemia y elevación de la VSG. En los linfomas agresivos es asimismo habitual la elevación de la lactatodeshidrogenasa y por lo común estos pacientes presentan parámetros manifiestos de desnutrición como, por ejemplo, hipoalbuminemia (un 86% de los casos en una serie previa).
En relación con el adenocarcinoma del intestino delgado, su manifestación clínica viene condicionada en la mayoría de los casos por el crecimiento intraluminal de estas lesiones. El síntoma más frecuente es el dolor abdominal, la distensión y las náuseas secundarias a un proceso obstructivo. La hemorragia gastrointestinal, generalmente crónica, es el segundo síntoma en frecuencia y se da en el 20–50% de los pacientes. Los tumores malignos del intestino delgado son raros en la población general, pero hay ciertos factores que predisponen a él, como los síndromes familiares polipósicos, la enfermedad de Crohn y la enfermedad celíaca. En pacientes con enfermedad celíaca el riesgo de adenocarcinoma del intestino delgado, un cáncer en general excepcional, está incrementado en comparación con la población general, aunque el riesgo global es bajo debido a la infrecuencia de este cáncer. Un reciente estudio poblacional ha estimado en 10 el incremento de riesgo de desarrollar un adenocarcinoma intestinal respecto a la población general27. Los tumores malignos del intestino delgado tienen en común con los del colon el hecho de que se considera que aparecen sobre un adenoma previo. A diferencia de lo que ocurre en la enfermedad de Crohn, en la que hay evidencia de una secuencia displasia-carcinoma, la enfermedad celíaca no evidencia una displasia adyacente a la mucosa tumoral. En la mayoría de las series de casos de pacientes con enfermedad celíaca y adenocarcinoma intestinal que se han publicado, a diferencia del predominio femenino que se observa en la celiaquía, los tumores son más frecuentes en el varón, con una mayor incidencia en la franja de edad de 49–70 años, y el riesgo de cáncer suele ser mayor en pacientes con enfermedad celíaca de larga evolución. Asimismo, en la mayoría de las series la localización predominante del carcinoma fue duodeno-yeyunal y excepcionalmente ileal28,29.
Los tumores neuroendocrinos representan el 1,2–1,5% de las neoplasias gastrointestinales. Su localización primordial es el recto (52%), el estómago (22%) y el intestino delgado (11%). Clásicamente los tumores neuroendocrinos se clasificaban sobre la base de su origen embrionario. Recientemente la Organización Mundial de la Salud ha establecido una nueva clasificación en función de su potencial maligno30. En esta clasificación, los tumores neuroendocrinos se dividen en tumores endocrinos bien diferenciados (con buen pronóstico y bajo potencial de malignidad), carcinomas endocrinos bien diferenciados (con alto grado de malignidad) y carcinomas endocrinos mal diferenciados (la mayoría son carcinomas de células pequeñas). El término «tumor carcinoide» se usa como sinónimo de tumor endocrino bien diferenciado, y «tumor carcinoide maligno» es sinónimo de carcinoma endocrino mal diferenciado. El intestino delgado, en particular el íleon, es el sitio donde con mayor frecuencia se presentan los tumores carcinoides, y suman el 30% de los carcinoides31. Dado que los tumores malignos del intestino delgado son infrecuentes, los carcinoides representan entre un 28 y un 38% de los tumores malignos. Estos tumores suelen aparecer a cualquier edad, pero predominan entre los 62 y los 65 años. Los síntomas clínicos más frecuentes son la obstrucción intestinal intermitente (46%) y el dolor abdominal (41%). La hemorragia, al igual que el síndrome carcinoide, es inusual en estos tumores32. Los tumores primarios generalmente permanecen asintomáticos, hasta que aparece una enfermedad voluminosa en los ganglios mesentéricos o hasta que desarrollan metástasis hepáticas con el síndrome carcinoide resultante (sofocos, diarrea, dolor abdominal, broncoconstricción). En la mayoría de los casos, en el momento del diagnóstico los tumores tienen un tamaño superior a 2cm, han invadido la muscular propia y condicionan una metastatización ganglionar local. Los tumores neuroendocrinos en esta localización pueden ser únicos o múltiples en un 40% de los casos. La mayoría de los pacientes con tumores carcinoides del intestino delgado comienzan con metástasis ganglionares o hepáticas, y un 5–7% de los casos presenta síndrome carcinoide33.
Los sarcomas gastrointestinales representan un 2% de los sarcomas de los adultos. El sarcoma más frecuente en el intestino delgado es el leiomiosarcoma (un 75% de los sarcomas gastrointestinales). La edad media de presentación de los sarcomas es de 60 años, con un ligero predominio del sexo masculino. Prácticamente la mitad de los sarcomas intestinales son de localización yeyunal (47%), y se localizan en el íleon y el duodeno en el 28 y el 25%, respectivamente. En relación con su estadio en el momento de la presentación, un 42% de los casos son tumores localizados y un 31% presenta metástasis a distancia34. Es un tumor que rara vez requiere la resección adenopática porque rara vez metastatiza a ganglios.
Los GIST, por su parte, son tumores mesenquimales frecuentes en el intestino delgado y constituyen el 20–45% de los casos malignos35. Se caracterizan por la expresión del receptor de membrana c-kit/CD117, que tiene actividad tirosincinasa y es sintetizado por el protooncogén c-kit36. El 60% de los GIST se localizan en la región gástrica, un 30% en el intestino delgado y un 5% en colon y recto. Su forma de presentación es muy variable. Los de pequeño tamaño (< 2cm) normalmente no producen síntomas y se detectan de forma incidental durante un estudio endoscópico o por pruebas de imagen. Los de mayor tamaño suelen manifestarse como consecuencia del efecto masa y desplazamiento por contigüidad. Las manifestaciones clínicas más habituales son la hemorragia —consecuencia de su efecto erosivo a la luz intestinal (40%)—, el efecto masa abdominal (40%) y el dolor (20%). Globalmente, dos tercios de los pacientes presentan hemorragia gastrointestinal, oculta o visible. La localización más frecuente es el yeyuno, seguida por la localización ileal y finalmente duodenal. Los GIST metastatizan con frecuencia al hígado, ocasionalmente a los ganglios linfáticos regionales36 y casi nunca a los pulmones, lugar donde con mayor frecuencia se diseminan los leiomiosarcomas y otros tipos de sarcomas tisulares (excepto los liposarcomas). Los factores de mal pronóstico o agresividad de estos tumores son el tamaño (>5–10cm) y el índice mitótico (más de 10 mitosis por 50 campos de gran aumento)37. Algunos estudios han observado que el lugar de origen confiere mal pronóstico a estos tumores, de modo que la localización intestinal tiene peor pronóstico que la localización gástrica38. No obstante, todos los GIST deben considerarse potencialmente malignos, con las posibles excepciones de los tumores de pequeño tamaño (< 1cm).
En conclusión, creo que la paciente está afectada de un tumor ileal maligno con extensión regional y a distancia. La localización focal no difusa y la falta de repercusión en los parámetros nutricionales y analíticos van en contra de un proceso linfomatoso, bien sea aislado o asociado a una enteropatía no diagnosticada, que creo que queda descartada a priori en esta paciente. El adenocarcinoma intestinal quedaría excluido por su afectación primordialmente duodenal proximal y su escasa frecuencia fuera del contexto de una enfermedad celíaca de base. Igualmente descartaríamos el leiomiosarcoma por su infrecuente diseminación ganglionar y por la ausencia de diseminación pulmonar, el lugar de diseminación más frecuente de este tumor. Finalmente, establecería el diagnóstico diferencial entre un carcinoma neuroendocrino diseminado sin síndrome carcinoide asociado y un GIST metastásico.
La exploración que debería efectuarse, desde mi punto de vista, es una punción aspirativa con aguja fina de las lesiones focales del hígado. Como segunda opción, realizaría una enteroscopia vía colónica, aunque la distancia del tumor respecto a la válvula ileocecal la hace más dificultosa. Si estas 2 opciones no dieran el diagnóstico, debería efectuarse una laparotomía exploradora.
Dr. Xavier Bosch. ¿La alta fiebre que presentaba la paciente le hace pensar más en un tumor neuroendocrino o en un GIST?
Dr. X. Bessa. Lo cierto es que no es un síntoma frecuente en ninguno de ellos, ya que en el tumor neuroendocrino lo que se describe con cierta frecuencia es la presencia de rubor.
Diagnóstico del Dr. X. BessaCarcinoma neuroendocrino diseminado o GIST metastásico.
Diagnóstico ClínicoNeoplasia de intestino delgado con metástasis hepáticas.
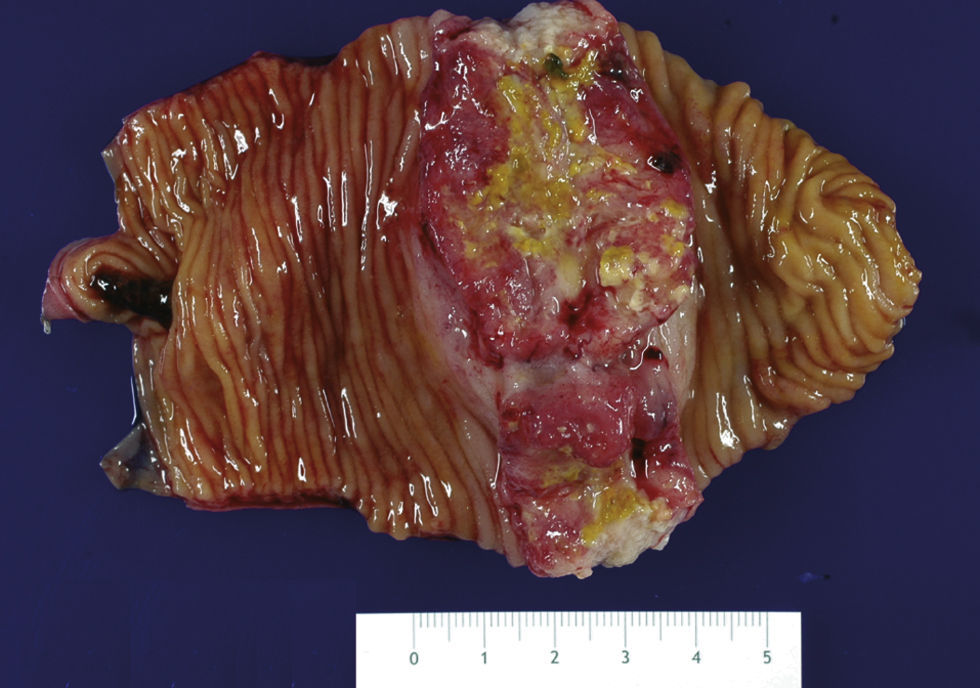
Discusión AnatomopatológicaProf. Josep A. Bombí. La prueba diagnóstica fue una laparoscopia exploradora en la que se observaron múltiples lesiones nodulares hepáticas, así como una tumoración estenosante yeyunal. Se efectuó una resección de un fragmento intestinal de 9,5cm y a la apertura se identificó una tumoración ulcerada de bordes sobreelevados que medía 3,7 por 3,5cm y ocupaba prácticamente toda la circunferencia (fig. 4). Distaba 2,2 y 5cm de los márgenes de resección, y a la sección presentaba un grosor máximo de 1,1cm, sobrepasaba la muscular y llegaba a menos de 0,2cm de la serosa.

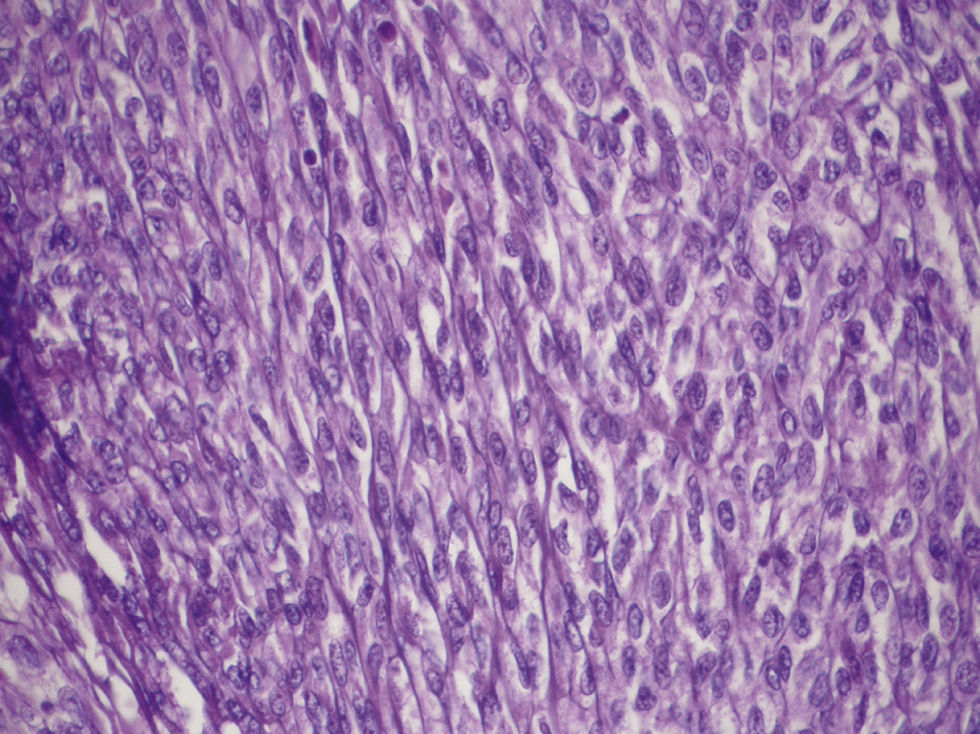
En el estudio histológico se apreció una neoplasia sólida de aspecto multinodular, más o menos fusocelular y en algunas áreas de aspecto epitelioide. Las células tenían un citoplasma bastante abundante, a veces claro, y los núcleos eran redondeados u ovalados, con nucléolos poco evidentes. Presentaba discreta atipia y abundantes figuras de mitosis, más de 5 por 50 campos de gran aumento. La actividad proliferativa medida con Ki-67 mostró más del 20% de células positivas (fig. 5).
")
Hace algunos años hubiéramos catalogado esta tumoración como neoplasia de músculo liso, leiomioma o leiomiosarcoma o leiomioblastoma. Con la incorporación de la microscopia electrónica y posteriormente de la inmunohistoquímica, hoy sabemos que estos tumores corresponden a un amplio abanico de neoplasias de distintos orígenes con un fenotipo semejante. El primer diagnóstico que debe considerarse es el de GIST, ya que son los tumores mesenquimales más frecuentes en el estómago e intestino delgado. Sus características histológicas son poco específicas, si bien esto ha cambiado con la incorporación de las nuevas técnicas de diagnóstico. Generalmente son de hallazgo ocasional por endoscopia, radiología o cirugía, o en ocasiones tras estudios por presencia de una masa abdominal, dolor abdominal o hemorragia gastrointestinal. Para su diagnóstico es muy importante el estudio inmunohistoquímico, ya que estos tumores son positivos para CD117 (proteína c-kit) en más del 95% de los casos y para CD34 (antígeno de la célula progenitora hematopoyética) en cerca del 70%39–41. Se ha propuesto el origen tumoral a partir de las células intersticiales de Cajal (células marcapasos del tubo digestivo que regulan su motilidad espontánea). Generalmente las células neoplásicas son negativas para actina de músculo liso (un 20–30% positividad) y para S100. Es muy importante el diagnóstico correcto de esta neoplasia, ya que en muchos casos tiene una buena respuesta terapéutica con imatinib, que es un inhibidor del receptor de la tirosincinasa42.
En nuestro caso, las tinciones inmunohistoquímicas para c-kit y CD34 fueron negativas, al igual que para enolasa neuronal específica, cromogranina, actina de músculo liso, citoqueratinas de amplio espectro, citoqueratina Cam 5/2, HMB-45, Melan A, alfafetoproteína y desmina. Por el contrario, se encontró positividad difusa para vimentina, anticuerpos antiendomisio, alfa-1-antitripsina, bcl-2, y positividad focal para S100, sinaptofisina, neurofilamentos y CD56.
Se ha encontrado que en un 4% de los casos los GIST son negativos al estudio inmunohistoquímico para c-kit y CD34, por lo que para descartarlos con seguridad se debe proceder al estudio genético43. Los GIST presentan mutaciones de c-kit en el 80–85% de los casos. El c-kit es un receptor con actividad tirosincinasa, codificado por un gen localizado en el cromosoma 4, que tiene 21 exones. Las mutaciones más frecuentes son en los exones 11 (75%), 9 (15%), 13 (5%) o 17 (5%). Parece que las mutaciones del exón 11 tienen un mejor pronóstico. Un pequeño porcentaje de GIST, especialmente los de localización gástrica, que no presentan alteraciones de c-kit tiene alteraciones en PDGFR-alfa (receptor alfa del factor de crecimiento derivado de plaquetas). El gen que codifica esta proteína se encuentra en el cromosoma 4, en una localización cercana al c-kit. Las mutaciones se encuentran en los exones 12 (1%), 18 (4%) o 14. En nuestro caso, el análisis genotípico mediante secuenciación directa de la muestra de ADN extraído de material parafinado (efectuado gracias a la colaboración del Dr. X. Matias Guiu) no evidenció ninguna mutación en los exones 9, 11, 13 y 17 del gen c-kit, ni en los exones 12, 13 y 18 del gen PDGFR-alfa. Por lo tanto, podemos descartar que se trate de un GIST. Además, los GIST aparecen a cualquier edad, pero el 75% en mayores de 50 años, lo que tampoco se corresponde con la edad de nuestra paciente.
Dentro del diagnóstico diferencial deberemos valorar la posibilidad de un sarcoma de origen en el músculo liso, en el sistema nervioso periférico (schwannoma, GANT —tumor gastrointestinal de nervios autónomos—, tumor de células granulares, etc.) o de otro tipo (epitelial, alveolar, sinovial, etc.), una fibromatosis mesentérica o un seudotumor fibroso reactivo nodular44,45, o una metástasis de otro origen, como un melanoma o un carcinoma. Con la morfología y la inmunohistoquímica del caso, podemos descartar con total seguridad que se trate de una neoplasia de origen epitelial, muscular, neuronal o neuroendocrina, de una fibromatosis o de un sarcoma epitelioide o alveolar.
Morfológicamente, una posibilidad es el sarcoma sinovial, ya que además por regla general aparece en personas más jóvenes. Este tumor tiene característicamente una translocación específica t(X;18) (p11;q11) que afecta a más del 90% de los casos. El estudio de hibridación in situ fluorescente (efectuado gracias a la colaboración del Dr. De Álava) para sarcoma sinovial fue negativo.
La microscopia electrónica de las células del presente caso demostró que tenían un citoplasma bastante mplio, con retículo relativamente abundante y presencia de glucógeno, sin evidenciar estructuras valorables de uniones de membrana, salvo pequeñas uniones densas que pueden aparecer en todo tipo de células. No encontramos filamentos de tipo intermedio que apuntasen a queratinas ni haces de filamentos de actina. Tampoco se observaron imágenes de gránulos de neurosecreción ni microtúbulos. Por todo ello, desde el punto de vista ultraestructural, no pudimos orientar ningún tipo de diferenciación epitelial, nerviosa o neuroectodérmica ni muscular. Tampoco observamos imágenes importantes de lisosomas para pensar en tumores fibrohistiocitarios.
Por todo ello, nos encontramos ante una neoplasia maligna de tipo mesenquimal en la que no podemos precisar ningún tipo de diferenciación, por lo que nos quedamos con el diagnóstico de sarcoma sin otra especificación (sarcoma NOS, de no other specification) de alto grado. En general, la ausencia de diferenciación en una neoplasia causa cierta desazón al pensar que quizá no ha sido bien estudiada, cosa que creemos no es el caso. Sin embargo, en los servicios de anatomía patológica, aunque es bastante frustrante, ocurre en ocasiones. La revisión que Loughrey et al46 publicaron en 2006 ejemplifica la dificultad de estos casos: de 37 casos de posible GIST que le fueron remitidos en consulta, tras su revisión se aceptaron sólo 26 como tales y, de los 11 restantes, 8 se diagnosticaron de sarcoma NOS al no encontrarse tampoco ninguna diferenciación específica. De los primeros, el 92% mostró respuesta positiva frente al imatinib y, en cambio, 3 pacientes del segundo grupo no obtuvieron ningún resultado con el mismo tratamiento.
Dr. Ricard Cervera. ¿Pueden comentarnos los médicos que atendieron a la paciente cuál ha sido su evolución?
Dr. Josep Maria Grau. Tras el diagnóstico, se consultó al servicio de oncología para decidir el tratamiento a seguir. Sin embargo, dado que llamaban la atención la fiebre tan elevada y los valores altos de reactantes de fase aguda, se solicitó un estudio de citocinas. En dicho estudio se hallaron valores muy elevados de las interleucinas 6 y 8 (por encima de 50 veces el límite superior de la normalidad), en tanto que la interleucina 1β y el factor de necrosis tumoral fueron normales.
Dr. Joan Maurel. Lo primero que quiero destacar es que la respuesta del GIST es muy diferente de la de los sarcomas, ya que el primero responde bien al tratamiento con imatinib. Por ello, mientras se esperaba el diagnóstico definitivo de anatomía patológica que, como ha relatado el Prof. Bombí, se alargó en el tiempo, debido a dicha dificultad en el diagnóstico patológico y a la edad de la paciente se realizó de forma excepcional tratamiento con imatinib durante un mes y medio, sin conseguir respuesta clínica. Cuando el diagnóstico de sarcoma NOS fue firme, se retiró dicho fármaco y se inició tratamiento con adriamicina en monoterapia, ya que se ha visto que su asociación con otros quimioterápicos no aporta ventajas clínicas. La fiebre desapareció por completo a las pocas semanas de iniciado dicho tratamiento y a día de hoy, tras 6 tandas de adriamicina, la paciente se encuentra en remisión parcial desde el punto de vista radiológico.
Diagnóstico AnatomopatológicoSarcoma intestinal sin otra especificación (sarcoma NOS) de alto grado.
Agradezco la colaboración de todos los miembros del Servicio de Digestivo del Hospital del Mar por su excelente valoración crítica del caso, al igual que la de los compañeros del Servicio de Radiología, en especial los Dres. Marcos Busto y Juan Sánchez.
Conferencia celebrada el 9 de abril de 2008 en el Hospital Clínic de Barcelona.
