




Hereditary hemorrhagic telangiectasia (HHT), also known as Rendu–Osler–Weber syndrome, is a rare autosomal dominant genetic disorder with an estimated prevalence ranging from 1 to 10,000.1 It is characterized by recurrent epistaxis, mucosal telangiectasias (include: lips, nose, moth, fingers, etc.), visceral arteriovenous malformations (ANMs) predominantly in the lungs, liver, and brain, and gastrointestinal telangiectasia with or without bleeding. Epistaxis affects more than 95% of HHT patients.2
In 1994, ENG, which is located on chromosome 9q34 and encoding for the protein endoglin (CD105), was the first gene identified in which mutations resulted in HHT, so HHT caused by ENG mutations is known as HHT type 1 (HHT1).3 Endoglin is a cell-surface glycoprotein that functions as part of the transforming growth factor beta (TGF-β) signaling complex that plays an important role in angiogenesis and vascular remodeling.3 HHT type 2 results from mutations in ACVRL1, which encodes the activin receptor-like kinase 1 (ALK1) located on chromosome 12.4 HHT3 and HHT4 mutations are situated on chromosomes 5q31 and 7q14 respectively, with no specific genes identified yet. Bone morphogenetic protein 9 (BMP9) also known as growth differentiation factor 2 (GDF2), encoded by BMP9 (also called GDF2), is a ligand for the ACVRL1 gene product ALK1. Consequently, mutations in BMP9/GDF2 result in the clinical manifestations of HHT and are referred to as HHT5.3 SMAD4 mutations caused both juvenile polyposis (JP) and HHT, called syndrome of juvenile polyposis and HHT (JP-HHT).5 This study aims to report a novel frameshift mutation deletion of the endoglin gene and investigate the genetic cause in the HHT family.
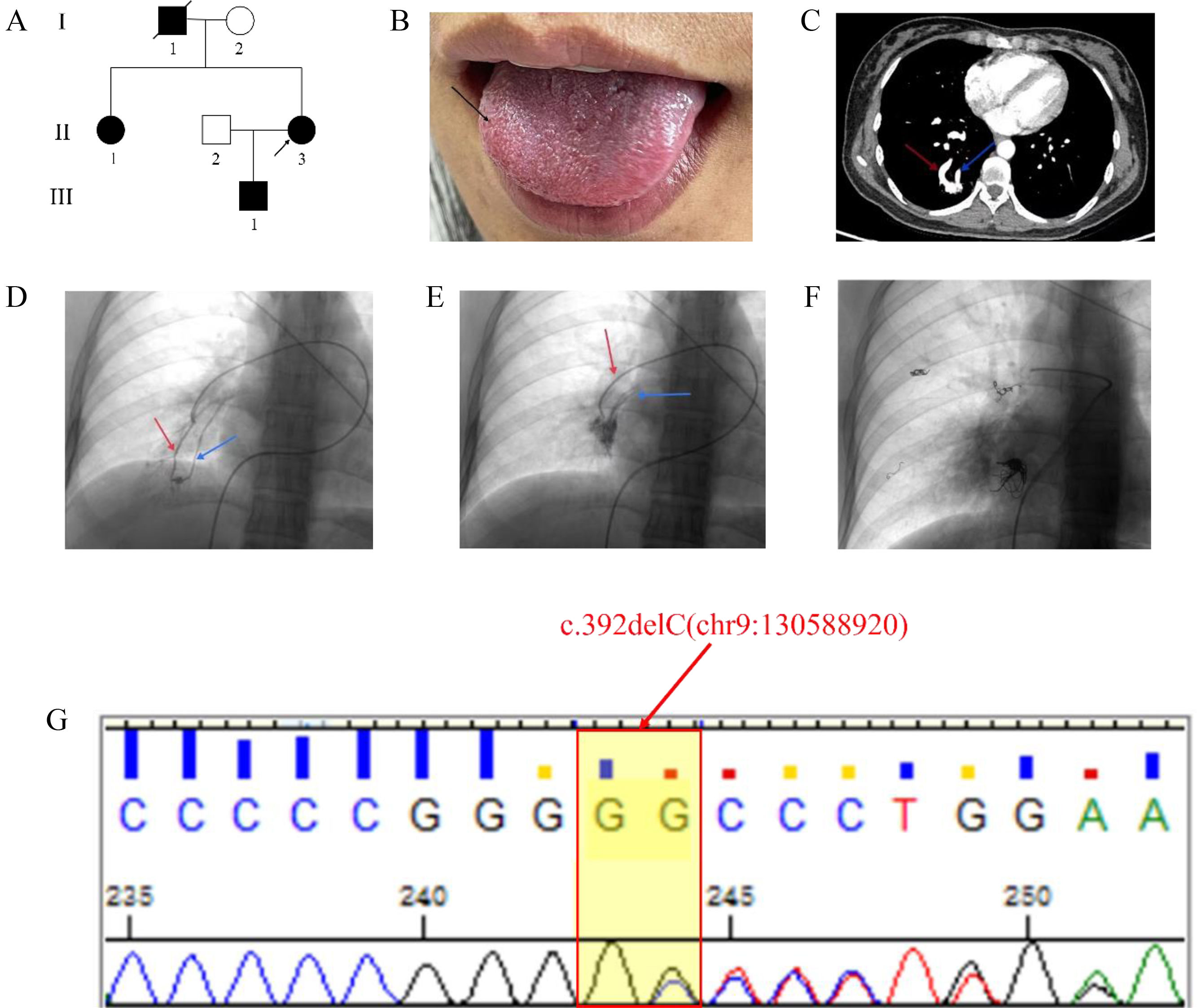
A 32-year-old Chinese woman (II-2 in Fig. 1A) was admitted to our hospital (the First Affiliated Hospital of Dali University) due to massive hemoptysis. The hemoptysis presented as bright red blood and lasted for approximately 10min, with an estimated total volume of 500–600ml. She had experienced recurrent epistaxis 10 years ago and did not receive regular treatment until now. On visual inspection, the patient exhibited multiple red spots on the tongue (Fig. 1B). Echocardiography did not show any obvious abnormalities. Arterial blood gas analysis indicated that the patient have type I respiratory failure with hypoxemia. Chest contrast-enhanced CT scan showed varying degrees of pulmonary arteriovenous malformation (Fig. 1C).
 Pedigree of the HHT family. The proband is indicated by an arrow. I, II, III: first, second, and third generation, respectively. (B) The proband (II-3) exhibited multiple red spots on the tongue (black arrow). (C) Chest contrast-enhanced CT scan showed arteriovenous malformations of the lungs. (D, E) Pulmonary angiography revealed multiple PAVM located in the inferior lobe area of the right lung. (F) Chest fluoroscopy after PAVM closure. (G) Sanger sequencing showed the patient had a heterozygous c.392delC (chr9:130588920) mutation. The red arrow shows the feeding artery and the blue arrow shows the draining vein; CT, computed tomography.")
The genetic and clinical characteristics of the family with HHT. (A) Pedigree of the HHT family. The proband is indicated by an arrow. I, II, III: first, second, and third generation, respectively. (B) The proband (II-3) exhibited multiple red spots on the tongue (black arrow). (C) Chest contrast-enhanced CT scan showed arteriovenous malformations of the lungs. (D, E) Pulmonary angiography revealed multiple PAVM located in the inferior lobe area of the right lung. (F) Chest fluoroscopy after PAVM closure. (G) Sanger sequencing showed the patient had a heterozygous c.392delC (chr9:130588920) mutation. The red arrow shows the feeding artery and the blue arrow shows the draining vein; CT, computed tomography.
On the day of admission, the patient he patient underwent transcatheter pulmonary arteriovenous malformation embolization (Fig. 1D–F). The patient's family history revealed that her father (I-1 in Fig. 1A) died of cerebral arteriovenous malformation 6 years ago, her sister (II-1 in Fig. 1A) also had frequent epistaxis, and there were arteriovenous malformations of the lungs and brain. The son is still young and has no abnormal clinical manifestations. Therefore, to investigate potential genetic factors contributing to the patient's recurrent epistaxis and arteriovenous malformation, mutation screening of the patient was performed by WES, and we identified a novel mutation c.392delC of ENG (Fig. 1G). Sanger sequencing showed the presence of the mutation in her sister and son. According to the American College of Medical Genetics and Genomics (ACMG) guideline criteria, the mutation was not present in the Human Gene Mutation Database (HGMD), Exome Aggregation Consortium (ExAC) database, the Genome Aggregation Database (gnomAD), ClinVar database and 1000 Genomes Project (TGP) database, and has not been reported in any published literature (PM2); and this mutation resulted in a substitution of proline with arginine at position 131 within the encoded peptide chain, leading to the creation of a stop at position 32 in the new reading frame [p.Pro131Argfs*32], which is predicted cause loss of normal protein function through either protein truncation or nonsense mediated mRNA decay (PSV1); the disease associated with this mutation corresponds to the clinical phenotype of the patient (PP4), in summary, the combination of evidence for this variant is PVS1+PM2+PP4,which is classified as “likely pathogenic”. Based on these findings, we ultimately diagnosed HHT in the patient, her sister and her son in according to the Curacao criteria.4
When the ENG gene is mutated, it can lead to the occurrence of diseases related to HHT vascular dysfunction, including recurrent epistaxis mucocutaneous telangiectasia, and arteriovenous malformation. And symptoms of HHT can vary widely depending on the location and severity of the telangiectasias and AVMs. While there is currently no cure for HHT, treatments are available to manage symptoms and prevent complications. Endoglin has distinct expression profiles in the pulmonary vasculature and is only co-expressed in the distal (pre-capillary) arteries, distal veins and capillaries, this is consistent with the tendency for pulmonary AVMs to form in the distal pulmonary vessels in HHT.1 In this study family, the proband's lung CT revealed a right lower pulmonary arteriovenous malformation, while cardiac color ultrasound indicated a normal diameter of the pulmonary artery root. These findings are in line with the expression characteristics of the ENG gene in pulmonary blood vessels. The location of pulmonary arteriovenous malformation in patients with HHT is variable and may not always result in serious clinical symptoms. In clinical practice, it is crucial for patients with arteriovenous malformations of internal organs to remain vigilant and prioritize screening for this condition.
In this study, we identified a previously unreported frameshift deletion mutation (c.392delC, p. Pro131Argfs*32) in the ENG gene. This novel finding not only enhances our understanding of the pathogenic factors and treatment strategies for HHT, but also broadens our knowledge of ENG gene mutations in HHT, providing fresh perspectives for future disease research.
Ethical statementsThe ethics committee of the First Affiliated Hospital of Dali University approved the research protocol, and written informed consent was obtained from every participant.
FundingThe study was supported by the Dali Bai Autonomous Prefecture Science and Technology Plan Project Key R&D Special Foundation, China [Numbers 20232903B03008] and 2020 Yunnan Province Local Undergraduate University Found Research Joint Project [Numbers 202001BA010001-047].
Informed consent statementInformed consent was obtained from all subject involved in the study.
Conflicts of interestThe authors declare no conflict of interest.
