





La enfermedad ósea de Paget se caracteriza por la alteración, en una o varias localizaciones óseas, del equilibrio entre formación y resorción ósea. Este desequilibrio da lugar a un hueso ensanchado, desorganizado, en muchos casos con una densidad ósea aumentada, aunque más frágil. Existiría una predisposición genética para su desarrollo, que explicaría entre un 5 y un 40% de los casos, sobre la que actuarían distintos factores ambientales. La enfermedad ósea de Paget fue considerada clásicamente la segunda enfermedad metabólica ósea más frecuente. Sin embargo, durante las últimas décadas presenta un marcado descenso tanto de la incidencia como de la gravedad clínica, lo que ha llevado a especular sobre la disminución o desaparición de la influencia de algún factor ambiental. Este descenso en la incidencia no debe servir como excusa para el abandono de su estudio, sino ser la razón para tratar de entender mejor su patogenia.
Paget's disease of bone is characterized by the alteration, in one or several bone locations, of the equilibrium between bone formation and bone resorption. This imbalance results in a disorganized, widened bone, in many cases with increased bone density, although more fragile. A genetic predisposition for Paget's disease of bone could explain between 5% and 40% of the cases. Different environmental factors should explain the rest of the cases. Paget's disease of bone was classically considered the second most common metabolic bone disease. However, in recent decades there has been a marked decrease in both incidence and clinical severity. These changes have led to believe that the influence of some environmental factor may have diminished or even disappeared. This decrease in incidence should not be an excuse for abandoning Paget's disease of bone research, but rather it should be the reason to remain searching to try to understand better its pathogenesis.
La enfermedad ósea de Paget (EOP) es la segunda enfermedad más frecuente del metabolismo óseo tras la osteoporosis. La enfermedad se caracteriza por la aparición de focos óseos en los que se aprecia un desequilibrio entre la formación y la resorción ósea, lo que acelera los procesos de reemplazo óseo. Esta alteración da lugar a unas zonas de hueso deformadas, con un patrón desorganizado, un aumento de la vascularización y una mayor fragilidad a pesar de su aparente aumento de densidad mineral ósea.
La enfermedad fue descrita por primera vez por Sir James Paget en 1877, con el nombre de osteitis deformans1. En la actualidad muchos de los pacientes con EOP son detectados de forma incidental al realizarse técnicas de imagen donde se aprecian las lesiones óseas, o por la detección de una elevación aislada de fosfatasa alcalina (FA). El dolor, la deformidad del hueso, la artrosis, las fracturas o la sordera son los síntomas y signos más frecuentes2.
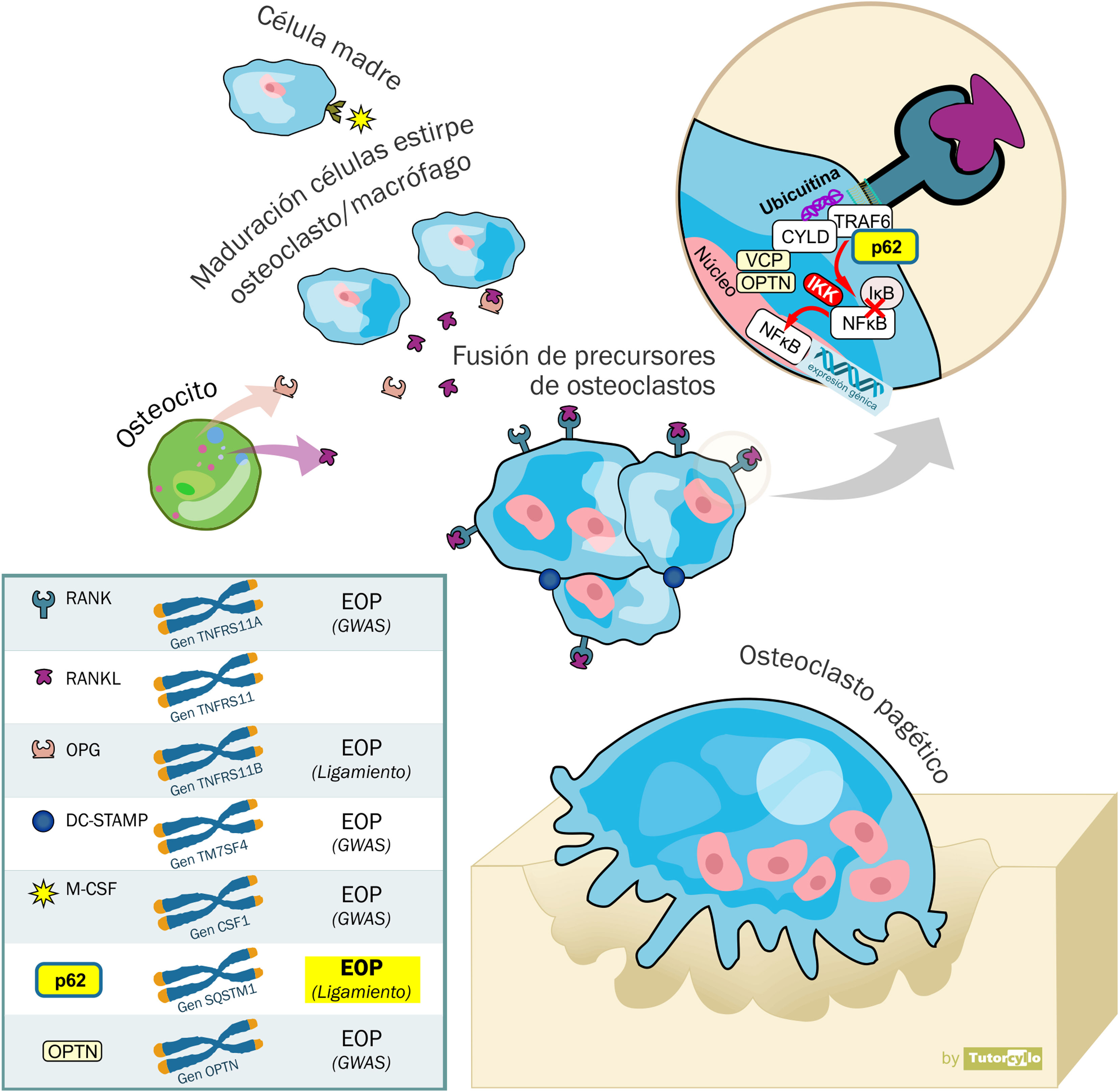
EtiopatogeniaPatología celularEl hueso está en constante remodelación con un acoplamiento entre las funciones de resorción y formación ósea. Los osteoclastos (derivados de precursores de la estirpe de los macrófagos) son responsables de la resorción ósea. Tras su acción, los osteoblastos (derivados de precursores de la estirpe de células madre mesenquimales) formarán nuevo hueso. Alguno de estos osteoblastos queda incluido en la matriz ósea, transformándose en osteocitos que tienen un papel en la regulación de la actividad de osteoblastos y osteoclastos mediante la producción de esclerostina y el receptor activator for nuclear factor κ B ligand (RANKL, «ligando del receptor activador del factor nuclear kappa B»).
En la EOP encontramos unos osteoclastos aumentados en tamaño y número y con más núcleos que los osteoclastos normales, con una mayor resistencia a la apoptosis3. Son más activos y presentan unas inclusiones nucleares4 de las que desconocemos su función, habiéndose especulado un origen viral, aunque en la actualidad se postula que podrían ser agregados de proteínas no degradadas por defectos en la autofagia5. En esta vía de activación participan proteínas que también se ven involucradas en los procesos de autofagia, lo que indica que puede ser una alteración de esta vía lo que da lugar a las alteraciones de los osteoclastos de la EOP. La más conocida de todas ellas, por estar además asociada a un gen relacionado con la susceptibilidad a padecer EOP es la p62. Esta proteína p62 tiene un papel crucial al reclutar proteínas ubiquitinadas para ser eliminadas en el autofagosoma. En los modelos animales de EOP, los osteoclastos pagéticos presentan niveles de p62 aumentados6. También la optineurina (OPTN) o la valosin-containing protein (VCP, «proteína contenedora de valosina») desempeñan un papel en la autofagia. Aún no conocemos si estos defectos en la autofagia son la causa directa del aumento en la formación de los osteoclastos o son simplemente un fenómeno colateral debido a la implicación de las proteínas involucradas tanto en la osteoclastogénesis como en la autofagia (p62, OPTN, VCP).
Predisposición genéticaLa primera descripción de la EOP como una posible enfermedad hereditaria por su alta prevalencia de casos familiares se publicó en 19497. La presencia de esta agregación familiar se cuantifica en un riesgo entre 7 y 10 veces mayor de padecerla en los familiares de primer grado de pacientes afectos que en la población general8.Esta agregación familiar, junto con la rareza de la enfermedad en población no caucásica9 y la presencia de un aumento del número de casos de EOP en determinadas áreas rurales (focos de alta prevalencia)10, indica la existencia de una predisposición al desarrollo de la enfermedad marcada por factores genéticos.
En las últimas décadas se han identificado distintas mutaciones que pertenecen a loci cercanos a genes involucrados en la interacción entre las células del hueso (fig. 1). La identificación de estos genes ha sido posible mediante estudios de ligamiento genético y de genome-wide association study (GWAS, «asociación del genoma completo»).
 es clave para la diferenciación desde la célula madre de estirpe osteoclasto/macrófago al osteoclasto. La fusión de los precursores de osteoclastos para formar osteoclastos maduros requiere dendritic cell specific transmembrane protein (DC-STAMP, «proteína transmembrana específica de las células dendríticas». Para la diferenciación y activación del osteoclasto es necesaria la activación del receptor activator for nuclear factor κ B ligand (RANK, «receptor activador del factor nuclear kappa B»). La unión del ligando del receptor activador del factor nuclear kappa B (RANKL), liberado por los osteoblastos, con el RANK, presente en la membrana celular de los osteoclastos, da lugar a la activación del factor nuclear potenciador de las cadenas ligeras kappa en las células B (NFκB). Este permanece en su estado inactivo el citosol celular formando un complejo con la proteína inhibitoria IκBα. La unión de RANKL y RANK da lugar a la activación de la cinasa IκB (IKK). Esta cinasa fosforila la proteína IκBα lo que provoca la ubiquitinación y disociación del complejo NFκB+IκBα. El NFκB así activado entra en el núcleo, uniéndose al ácido desoxirribonucleico (ADN) y dando lugar a la activación del ácido ribonucleico (ARN) mensajero y la formación de proteínas que provoca un cambio en la función celular. RANK también está implicado en la regulación de la autofagia. La interacción con p62 es clave para la activación de NFκB, facilitando la traducción de la señal del RANK. El tumor necrosis factor 6 receptor (TRAF6, «factor 6 asociado con el receptor del factor de necrosis tumoral) y el CYLD (cilindromatosis conservada) participan en la activación. La osteoprotegerina (OPG) es una proteína soluble con una gran similitud estructural con el RANKL, también liberado por los osteocitos, y que compite con este en su unión con RANK regulando a la baja la activación del NFκB al evitar la unión RANKL-RANK. La valosin-containing protein (VCP, «proteína contenedora de valosina») y la optineurina (OPTN) participan en la regulación de la señalización del NFκB.")
Regulación de la fisiología del osteoclasto. El macrophage colony-stimulating factor (M-CSF, «factor estimulante de las colonias de macrófagos») es clave para la diferenciación desde la célula madre de estirpe osteoclasto/macrófago al osteoclasto. La fusión de los precursores de osteoclastos para formar osteoclastos maduros requiere dendritic cell specific transmembrane protein (DC-STAMP, «proteína transmembrana específica de las células dendríticas». Para la diferenciación y activación del osteoclasto es necesaria la activación del receptor activator for nuclear factor κ B ligand (RANK, «receptor activador del factor nuclear kappa B»). La unión del ligando del receptor activador del factor nuclear kappa B (RANKL), liberado por los osteoblastos, con el RANK, presente en la membrana celular de los osteoclastos, da lugar a la activación del factor nuclear potenciador de las cadenas ligeras kappa en las células B (NFκB). Este permanece en su estado inactivo el citosol celular formando un complejo con la proteína inhibitoria IκBα. La unión de RANKL y RANK da lugar a la activación de la cinasa IκB (IKK). Esta cinasa fosforila la proteína IκBα lo que provoca la ubiquitinación y disociación del complejo NFκB+IκBα. El NFκB así activado entra en el núcleo, uniéndose al ácido desoxirribonucleico (ADN) y dando lugar a la activación del ácido ribonucleico (ARN) mensajero y la formación de proteínas que provoca un cambio en la función celular. RANK también está implicado en la regulación de la autofagia. La interacción con p62 es clave para la activación de NFκB, facilitando la traducción de la señal del RANK. El tumor necrosis factor 6 receptor (TRAF6, «factor 6 asociado con el receptor del factor de necrosis tumoral) y el CYLD (cilindromatosis conservada) participan en la activación. La osteoprotegerina (OPG) es una proteína soluble con una gran similitud estructural con el RANKL, también liberado por los osteocitos, y que compite con este en su unión con RANK regulando a la baja la activación del NFκB al evitar la unión RANKL-RANK. La valosin-containing protein (VCP, «proteína contenedora de valosina») y la optineurina (OPTN) participan en la regulación de la señalización del NFκB.
La búsqueda de genes candidatos para explicar la etiología de la EOP mediante estudios de ligamiento en familias con EOP hereditaria tuvo su mayor éxito a principios del siglo con la publicación de un estudio que demostraba la presencia de mutaciones en el cromosoma 5q35 en familias francocandienses11 que fue replicado en familias del Reino Unido12. Este locus se relaciona con el gen del sequestosoma (SQSTM1) que codifica la proteína p62. En posteriores estudios se han detectado hasta otras 30 mutaciones relacionadas con el gen SQSTM1. Aunque las mutaciones en el gen del SQSTM1 siguen siendo en la actualidad la principal causa genética identificada de la EOP, su espectro no es universal y se detecta solo en un 25-40% de los casos familiares y en menos del 10% de los pacientes sin ellos13. Estas mutaciones en el gen del SQSTM1 se consideran mutaciones que afectan a las células madre de la línea germinal, es decir, que afectan a todas las células del organismo descendientes de la célula original mutada. Además de esta herencia genética germinal, también se han descrito mutaciones somáticas (mutaciones que solo se encuentran en células de los tejidos afectos), en el gen del SQSTM1 en casos de EOP esporádica14,15. Esto podría explicar en parte la naturaleza focal de la EOP.
Además de en la EOP, los estudios de ligamiento en otras enfermedades hereditarias relacionadas con el espectro de la EOP han demostrado mutaciones relacionadas con el metabolismo óseo. Entre ellas destacan: 1) la osteólisis expansiva familiar (OMIM 174810): gen TNFRSF11a asociado con alteraciones en la señal del RANK16, 2) el Paget juvenil (OMIM 239000): gen TNFRSF11b que codifica la osteoprotegerina17 y 3) el complejo de EOP, demencia frontotemporal y miopatía por cuerpos de inclusión (OMIM 167320): asociado con el gen de la VCP18. Los estudios de ligamiento en EOP no demostraron asociación en el caso de los genes del TNFRSF11a19 (aunque sí la demostrarían en los GWAS como se verá más adelante, y en estudios mediante polimorfismos de nucleótido único20) ni en VCP21, pero sí en el del TNFRSF11b22.
Los GWAS han identificado otros 7 loci adicionales que explicarían un 13% del riesgo en pacientes con EOP y antecedentes familiares que no presentan mutaciones en el gen del SQSTM123,24. Destacan los genes relacionados con la fisiología del osteoclasto: a) el gen CSF1, que codifica el factor estimulante de colonias de macrófagos, b) el TNFRSF11A, que codifica el RANK, c) el TM7SF4, que codifica la proteína transmembrana específica de las células dendríticas, d) el OPTN que codifica la optineurina o e) el RIN3, que codifica un factor de intercambio de guanina expresado por los osteoclastos (fig. 1). Los genes implicados en los otros loci identificados son menos claros. La presencia de alguno de estos 7 loci adicionales aumenta el riesgo de padecer la enfermedad entre el 1,4 y el 1,7. Por último, también se ha investigado la posible asociación de la EOP con otros mediadores de la actividad del osteoclasto como la citocinas proinflamatorias25.
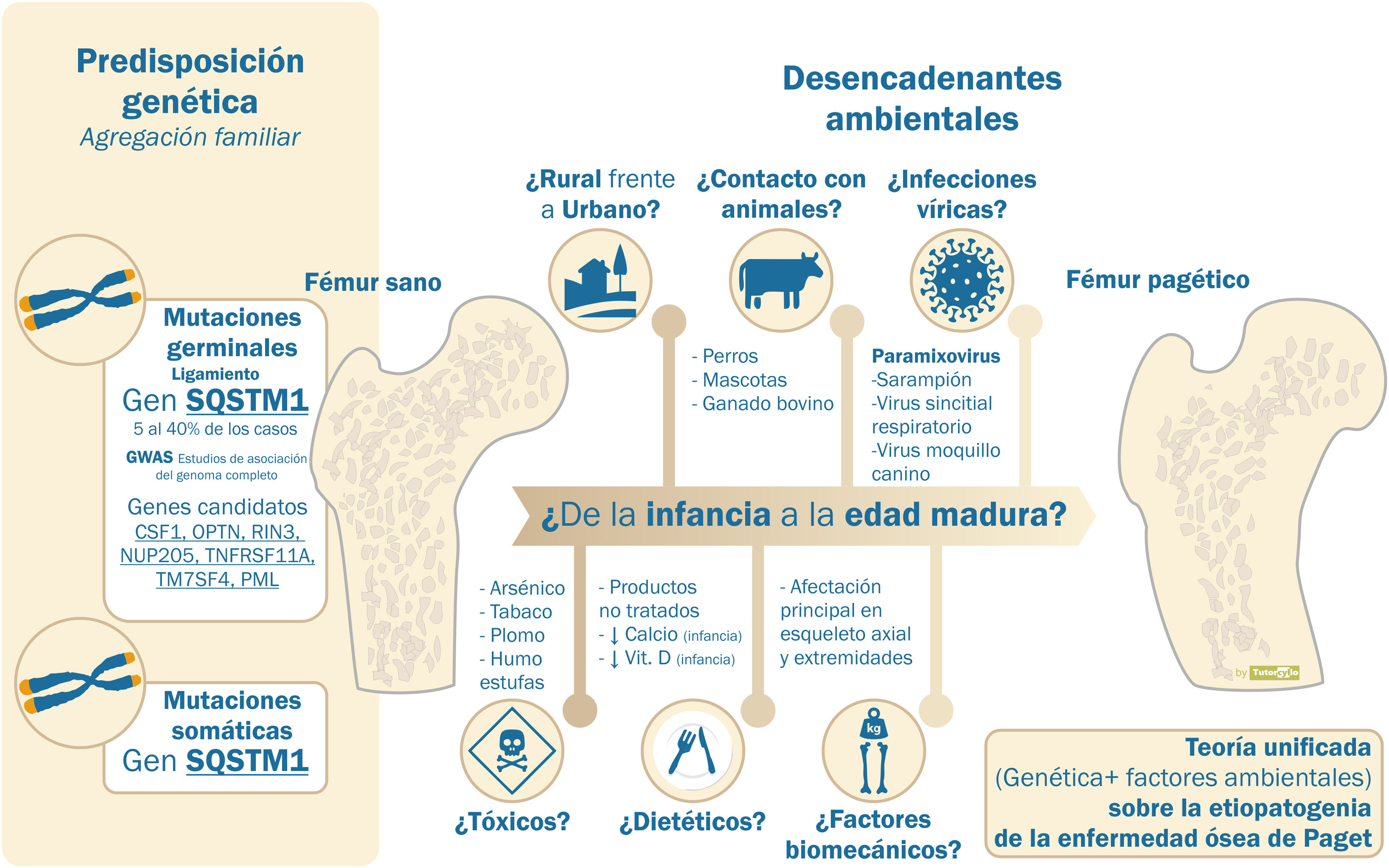
Sin embargo, el componente hereditario no es suficiente para explicar la enfermedad y existen argumentos que obligan a considerar la interacción con factores ambientales (tabla 1). El descenso de la incidencia y la gravedad descrito en las últimas décadas10 favorecería la teoría ambiental. Este descenso se ha demostrado incluso en pacientes portadores de mutaciones en el gen SQSTM1. Estos pacientes presentan en la actualidad un fenotipo más atenuado que su progenitores y tienen una edad de diagnóstico más tardía que estos26,27. El motivo de este retardo en la penetrancia genética es desconocido. La disminución en la exposición a factores ambientales se sugiere como mecanismo más probable para explicar estos fenómenos (fig. 2).
Etiología de la enfermedad ósea de Paget. Predisposición frente a influencia de factores ambientales
| Argumentos a favor de la predisposición genética | Argumentos a favor de la presencia de factores ambientales |
|---|---|
| • Historia familiar, con hasta un 40% de familiares afectos en algunas cohortes• Diferencias según el grupo étnico (prevalencia muy baja en pacientes asiáticos frente a caucásicos o africanos que viven en las mismas áreas)• Persistencia de tasas de prevalencia superiores en algunas poblaciones pese a los movimientos migratorios (persistencia de mayor tasa de casos en británicos emigrados a Australia frente a los locales)• Determinación de genes candidatos en estudios de ligamiento (especialmente gen SQSTM1/p62) | • Heterogeneidad de la presentación clínica de la enfermedad ósea de Paget, con afectación ósea focal• Rapidez de los cambios en la prevalencia y gravedad de la presentación clínica de la enfermedad en las últimas décadas• Demostración de asociación en los estudios de algunos factores como la sobrecarga mecánica de los huesos, la presencia de toxinas, la predominancia en el ambiente rural frente al urbano…• Reproducción de la enfermedad ósea de Paget en modelos de ratón y osteoclastos humanos utilizando virus del sarampión |

Etiopatogenia de la enfermedad ósea de Paget, teoría unificada. Son varios los genes que se han relacionado con la etiopatogenia de la enfermedad ósea de Paget. Sin embargo, este componente genético no puede explicar más allá de un 40% de las formas familiares y menos de un 10% de las formas esporádicas. La presencia de factores ambientales, aún no confirmados, ha de ser necesaria para el desarrollo de la enfermedad.
Son varios los posibles factores ambientales involucrados en la patogenia de la EOP aunque no existen evidencias claras de ninguno de ellos.
Uno de los que más documentación ha generado es la participación de infecciones víricas. La presencia de unas inclusiones en los osteoclastos que recordaban a partículas relacionadas con paramixovirus28 ha generado la hipótesis de la etiología vírica. Las evidencias sobre el origen de estas inclusiones son contradictorias, con la demostración inicial de partículas víricas en los osteoclastos29 o en células sanguíneas circulantes30 que no ha sido replicada en posteriores estudios31. No existe un acuerdo sobre su papel. Tampoco el análisis de la respuesta mediante anticuerpos a distintos virus (paramixovirus: sarampión, virus respiratorio sincitial, virus del moquillo canino, paperas, rubeola o varicela zóster) se ha asociado con la EOP o su gravedad32. La provocación de cambios similares a los de la EOP en modelos de laboratorio o de ratón con la infusión del virus del sarampión o su nucleocápside es un dato que apoya la teoría del origen vírico33.
Otros factores con los que se ha relacionado a la EOP son: 1) el contacto con animales como los perros34, no confirmado por otros investigadores35, 2) la residencia en el mundo rural36,37, 3) factores dietéticos como la baja ingesta de leche38 o la deficiencia vitamina D39 en la infancia, 4) la presencia de tóxicos como el arsénico40, el tabaco41 o el uso de estufas de madera en la infancia42, o 5) factores biomecánicos, al ser los huesos afectos principalmente aquellos relacionados con la carga, como en el caso del jugador de billar y la afectación pagética en la mano43.
En cualquier caso, la teoría que parece más factible para entender la complejidad de la etiopatogenia de la EOP es que, sobre una susceptibilidad genética, se asocie la participación de distintos factores ambientales, aún no confirmados, que den lugar al desarrollo de la enfermedad a lo largo de años. Esta teoría se conoce como teoría unificada. Estos factores ambientales, además de actuar sobre pacientes con una predisposición genética, también podrían dar lugar a cambios epigenéticos locales en otros pacientes sin dicha predisposición44. Estos cambios epigenéticos podrían justificar la presentación tan heterogénea de la enfermedad en cada paciente, aunque esto no ha sido aún demostrado. Por último, el papel de los microARN, pequeñas cadenas de ARN no codificadoras pero que regulan la expresión de los genes, incluidos los genes relacionados con el metabolismo óseo y la formación de los osteoclastos, también parece una futura línea de investigación45.
El estudio ZiPP46, donde se está siguiendo durante años a sujetos sanos portadores de mutaciones en el gen SQSTM1 que son familiares de pacientes con EOP, permitirá conocer más datos sobre el papel de la presencia de la predisposición genética asociada con las mutaciones en el SQSTM1 en el desarrollo de la enfermedad. En este estudio los sujetos sanos recibían de forma aleatoria la administración de una dosis de zoledronato o placebo al comienzo del seguimiento.
EpidemiologíaAl tratarse la EOP de una enfermedad ósea, y que por tanto deja una huella que perdura en el tiempo, es interesante referirse a los estudios arqueológicos para hablar de la epidemiología de la enfermedad. La elevada prevalencia de la enfermedad en el Reino Unido y en países con elevada proporción de población de origen anglosajón, como son las antiguas colonias de Australia o Nueva Zelanda, ha llevado a varios autores a proponer el origen de la enfermedad en las islas británicas. Este hecho parece confirmarse con un marcado aumento en el número de esqueletos con rasgos de la EOP procedentes del Reino Unido frente a otros países europeos47. Esta teoría es rebatida en un estudio español donde se hace un repaso de los casos paleopatológicos descritos de la enfermedad, estando los más antiguos descritos en yacimientos franceses, italianos o españoles, por lo que los autores postulan que la enfermedad no tuvo origen británico, sino que probablemente fue introducida en las islas durante la ocupación romana48. Además, estos autores describen casos hallados en yacimientos de la América precolombina, lo que hace más incierto cuál es el verdadero origen de la enfermedad.
La epidemiología de la EOP se ha caracterizado por una marcada heterogeneidad entre las distintas áreas del planeta, con un claro aumento de la prevalencia en las zonas de origen anglosajón y una escasa representación en Asia o África. Además de la marcada diferencia entre países, existen también una gran disparidad dentro de los propios países, con la presencia de los focos de alta prevalencia de la EOP en regiones donde el número de casos es mucho más elevado que en las regiones vecinas. El foco de alta prevalencia más conocido es el de Lancashire49 en Reino Unido. En Italia o España también se han descrito focos de alta prevalencia como el de Vitigudino, en Salamanca50, o el de la sierra de la Cabrera en Madrid51. Los focos de alta prevalencia españoles e italianos se caracterizan por corresponder a zonas rurales, escasamente pobladas y algo aisladas. Cuando se ha intentado explicar el origen de estos focos se ha pensado en la presencia de factores ambientales, entre ellos aquellos que tienen que ver con el contacto con animales. En el caso italiano existe una gran proporción de formas familiares lo que indica un origen genético.
Sin embargo, si algo hace interesante la epidemiología de la EOP no es su heterogeneidad, sino los marcados cambios que ha experimentado en las últimas décadas10, con un descenso extremo de la prevalencia, más marcado aún en los focos de alta prevalencia52. Este descenso ha ido acompañado de un retardo en la presentación de la enfermedad, que cada vez se diagnostica más tarde, y de un descenso en la gravedad de la presentación, con afectaciones esqueléticas menos extensas y valores de laboratorio menos elevados. Aunque se ha intentado explicar parte de estos cambios por las diferencias en el fondo genético debido a la importante inmigración desde zonas de baja prevalencia de EOP ocurrida durante las últimas décadas en países como el Reino Unido (desde la India o Pakistán) o España (desde los países de sur de América), lo cierto es que esta inmigración no ha afectado de forma importante a las regiones rurales como Vitigudino, que han sido las áreas donde realmente más se ha notado el descenso en la prevalencia de la EOP. La reducción en la exposición a algún agente ambiental aún no determinado, como podrían ser los cambios relacionadas con el control veterinario del ganado, podría explicar mejor los drásticos cambios en la prevalencia de la EOP.
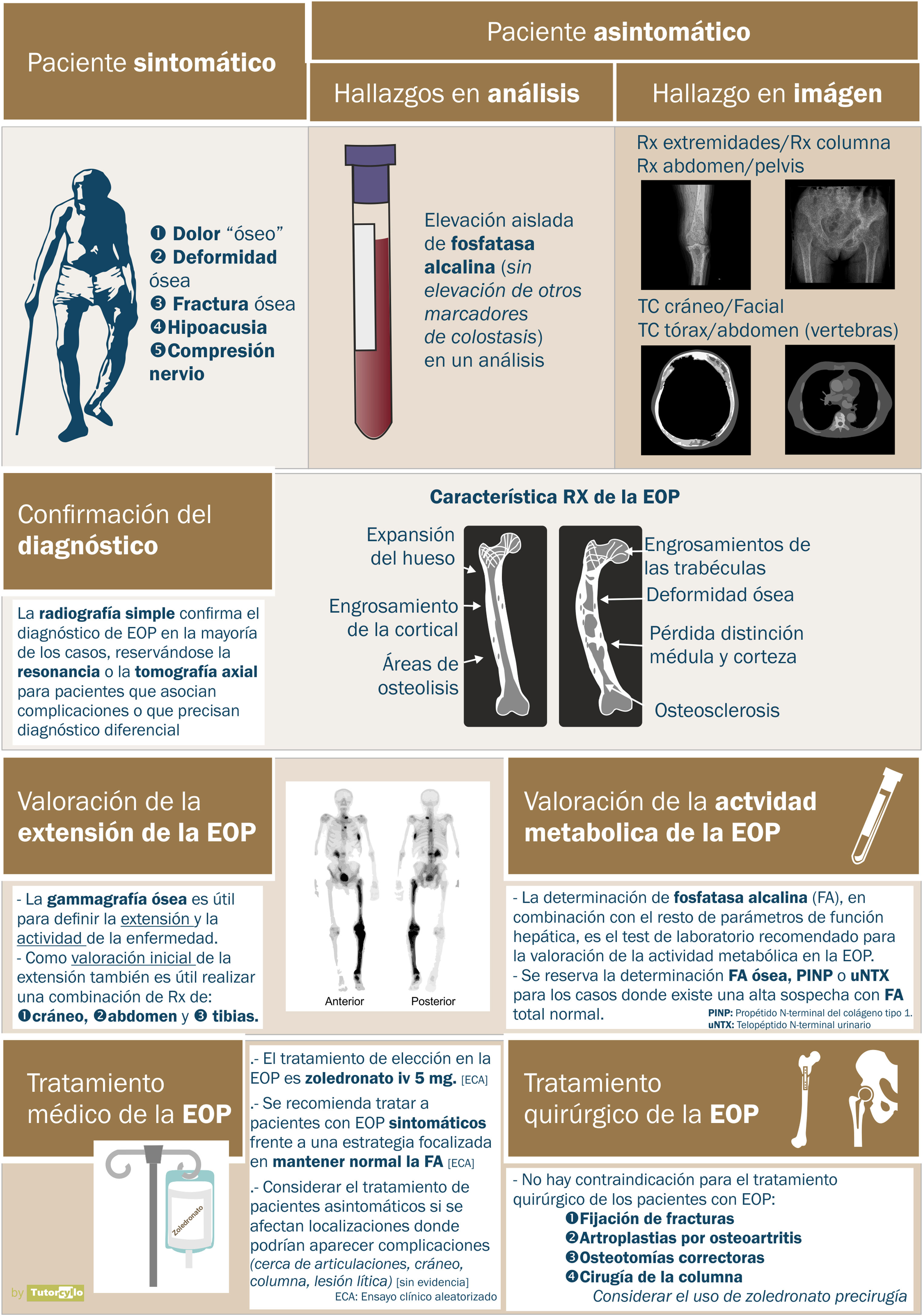
ClínicaEn una revisión sistemática publicada en 20142 se valoró la presentación clínica de la enfermedad. El principal síntoma de los pacientes con EOP fue el dolor en relación con la enfermedad, aunque la prevalencia de este síntoma varió de forma muy significativa entre las distintas series incluidas en la revisión (desde un 18% a un 92% de los casos), lo que hace difícil estimar una verdadera prevalencia de los síntomas. Estas diferencias también se confirman en nuestro medio. La sensación general es que la gran mayoría de los pacientes son asintomáticos, y sus diagnósticos de realizan de forma incidental al llevar a cabo otras pruebas diagnósticas (pruebas de imagen o determinación de FA). En una serie británica publicada recientemente, hasta un 74% de pacientes con EOP diagnosticados en atención primaria no eran referidos a consultas especializadas para valorar tratamiento53.
Además del dolor causado por la propia actividad local de la enfermedad, la deformidad que provoca esta da lugar a artrosis de las articulaciones lo que también genera dolor local. Diferenciar entre el dolor causado por la actividad de la enfermedad y el secundario a la artrosis resultante por la deformidad a la que ha dado lugar muchas veces es complejo y va dar lugar a la instauración de tratamientos antirresortivos que no van a mejorar la clínica del paciente.
La deformidad ósea también da lugar a trastornos neurológicos, por compresión de los nervios o por estenosis del canal medular, y la sordera, por alteración de las estructuras óseas que contiene el oído interno.
Las fracturas sobre los huesos afectos están también aumentadas al tratarse de huesos mal estructurados que resisten peor las cargas biomecánicas.
Manifestaciones clásicas como la insuficiencia cardíaca asociada al aumento de la vascularización del hueso, la hipercalcemia o la transformación neoplásica en osteosarcomas o tumores de células gigantes de las lesiones54 son en la actualidad infrecuentes o incluso inexistentes.
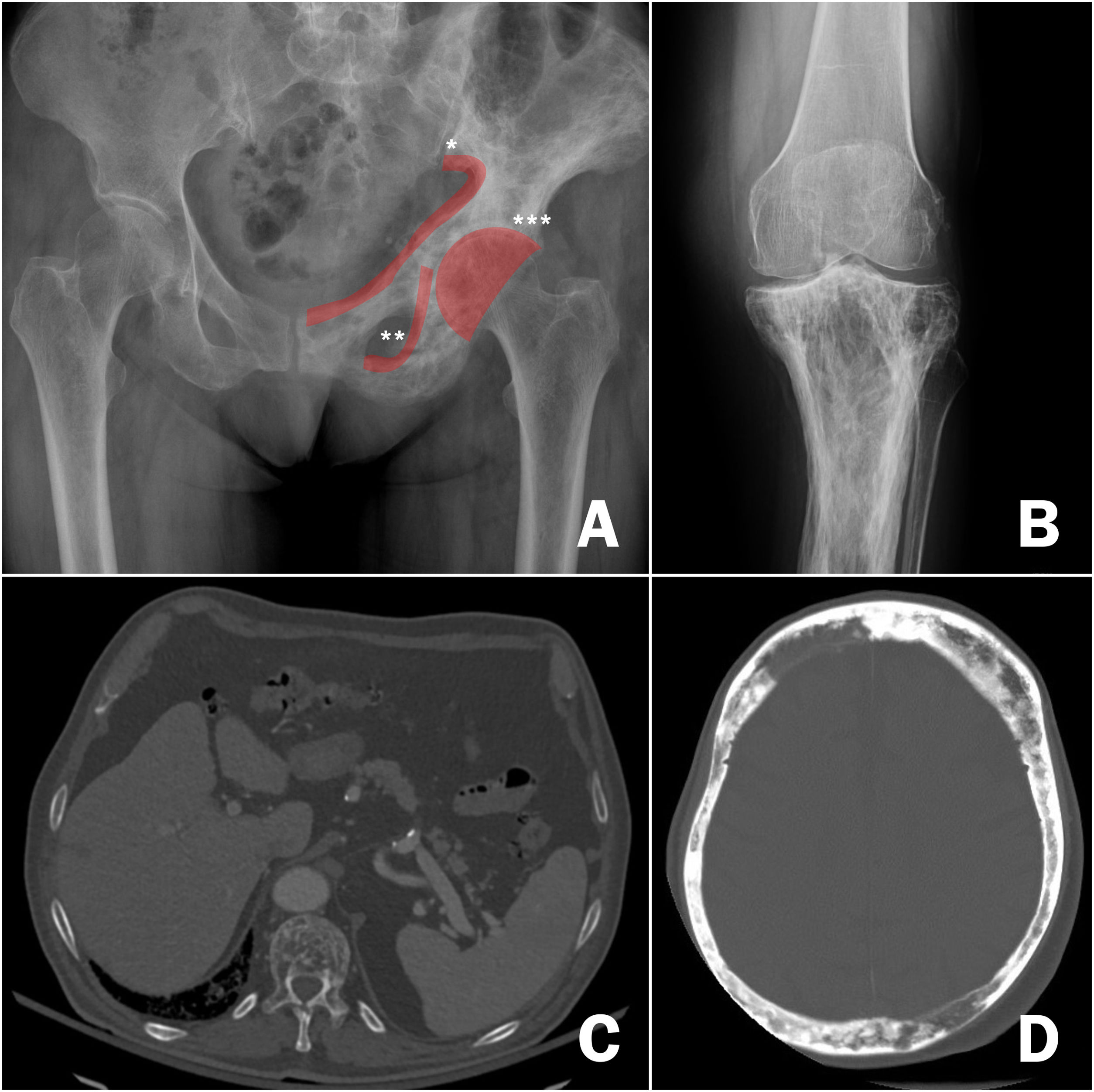
DiagnósticoUna vez establecida la sospecha de la EOP, es necesario confirmarla mediante la realización de pruebas de imagen que permitan demostrar los cambios propios de la enfermedad (fig. 3). En una primera fase de la afectación ósea, se describe un «frente lítico» o «Blade of Grass», con afectación de las corticales. Posteriormente, el hueso pagético se caracterizará por la presencia tanto de un engrosamiento de la cortical como de una trabeculación alterada y grosera que acaba provocando el agrandamiento y la deformidad del hueso (fig. 3B, comparación entre la cortical y las trabéculas del fémur sano y la tibia con EOP). Es característica la asociación de zonas osteoblásticas y osteolíticas en la enfermedad. En los casos de la afectación de la columna vertebral, las lesiones de la EOP se presentan con un aumento de la opacidad, igual que las metástasis osteoblásticas de algunos tumores. Sin embargo, la deformidad de la vértebra y el aumento del grosor de la cortical suelen hacer sencilla la diferenciación entre unas y otras. En otras localizaciones como la pelvis, también será relativamente sencillo diferenciar la afectación osteoblástica de la EOP de la metastásica de los tumores ya que en la EOP el hueso se agranda, se alteran las líneas ileopectina, ilioisquiática o se deforma el acetábulo (fig. 3A). En los casos de duda diagnóstica o ante la sospecha de complicaciones, es recomendable solicitar estudios diagnósticos de imagen adicionales como la tomografía axial computarizada o la resonancia magnética. Solo en casos en lo que pese a estas pruebas de imagen no fuera posible establecer el diagnóstico de la enfermedad se solicitaría una biopsia ósea. En la práctica habitual es realmente infrecuente necesitar un estudio anatomopatológico para la confirmación de la EOP.
 afectación de la hemipelvis izquierda, con alternancia de imágenes osteolíticas y osteoblásticas y engrosamiento de las corticales, *línea ileopectina, ** línea ilioisquiática, *** acetábulo. B) Afectación de la región superior de la tibia, con un engrosamiento marcado de las corticales y alteración de las trabéculas en la región superior. C) Afectación de vértebra dorsal en tomografía axial computarizada pulmonar, destaca la alteración de la estructura del cuerpo vertebral. D) Afectación craneal en tomografía axial computarizada craneal solicitada por ictus. Marcado engrosamiento de la cortical.")
Imágenes radiológicas de enfermedad ósea de Paget. A) afectación de la hemipelvis izquierda, con alternancia de imágenes osteolíticas y osteoblásticas y engrosamiento de las corticales, *línea ileopectina, ** línea ilioisquiática, *** acetábulo. B) Afectación de la región superior de la tibia, con un engrosamiento marcado de las corticales y alteración de las trabéculas en la región superior. C) Afectación de vértebra dorsal en tomografía axial computarizada pulmonar, destaca la alteración de la estructura del cuerpo vertebral. D) Afectación craneal en tomografía axial computarizada craneal solicitada por ictus. Marcado engrosamiento de la cortical.
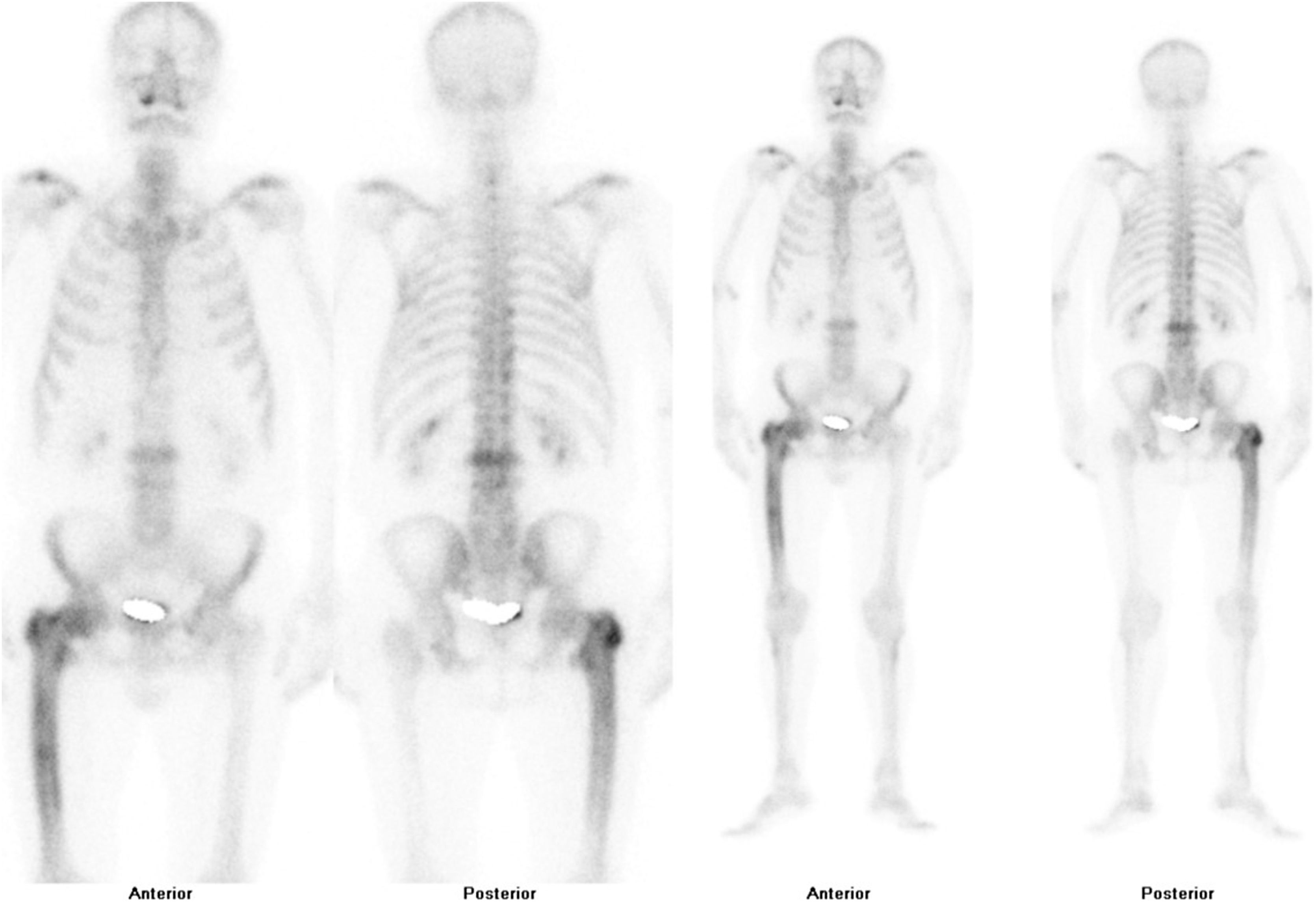
Tras la confirmación de la enfermedad en la radiografía simple o la tomografía axial computarizada, se completa el estudio de extensión de la enfermedad valorando la posibilidad de afectación en otras zonas del esqueleto. La gammagrafía ósea con tecnecio 99 (fig. 4) es la prueba más utilizada. Una alternativa sería la realización de una serie ósea radiológica que incluyese el cráneo y la cara, el abdomen y las tibias. Esta aproximación demostró una sensibilidad del 93% en una cohorte española de EOP55.

En cuanto a la valoración de la actividad bioquímica de la enfermedad, la determinación de FA es la prueba más estandarizada, con la detección de su elevación aislada dentro de las determinaciones de colestasis hepática. Solo en el caso de alta sospecha con normalidad de la FA podría ser de utilidad solicitar marcadores de recambio óseo adicionales como la fracción ósea de la FA o el propéptido aminoterminal de procolágeno tipo 1 o el telopéptido N terminal de colágeno tipo 1.
TratamientoLa principal indicación del tratamiento es el control del dolor local en relación con una zona afecta en los casos que esté relaciona con la actividad de la propia enfermedad. En estos pacientes, el uso de bisfosfonatos es eficaz para la reducción del dolor, con un número necesario de pacientes a tratar de 5, con intervalo de confianza entre 1 y 3556. El bisfosfonato de elección es el zoledronato por vía intravenosa, que se ha mostrado superior a otros bisfosfonatos como el risedronato57 o el pamidronato. Otros tratamientos como la calcitonina pueden ayudar al control del dolor, pero en la actualidad su uso se limita a pacientes con contraindicación para la administración de bisfosfonatos. Existen casos publicados de respuesta de la EOP con el uso de denosumab, otro fármaco antirresortivo, pero en la actualidad no está aprobado su uso en esta indicación.
En relación con el tratamiento con zoledronato por vía intravenosa, su administración puede provocar un cuadro seudogripal en las primeras 24 a 48h, con mialgias, cefalea o náuseas en casi uno de cada 4 pacientes durante la primera infusión, siendo menos frecuente en siguientes infusiones. Este cuadro es autolimitado. Algunos autores plantean el uso de paracetamol o dexametasona58 profilácticos en el día del tratamiento y siguientes. También será necesario conocer los niveles de vitamina D del paciente antes de su administración, ya que en un paciente con hipovitaminosis existe riesgo de hipocalcemia tras la administración del fármaco.
Aunque la indicación de tratamiento en paciente con EOP y dolor en la región afecta está establecida, la dificultad con la que nos encontraremos en no pocos pacientes será conocer cuál es el origen del dolor. Identificar si lo que lo provoca es la actividad de la propia EOP, u otras causas relacionadas con esta como la osteoartrosis condicionada por la deformidad ósea, las compresiones nerviosas relacionadas con la deformidad o las seudofracturas, no siempre será fácil. En estos casos, una historia y exploración clínica nos aportarán la información más adecuada para decidir el origen del dolor. La presencia de elevación en los marcadores de formación ósea, como es el caso de la FA, puede ayudar en la toma de decisiones, ya que la actividad de la enfermedad suele asociar valores elevados de FA, pero no debe dirigirla. En los casos en que el origen del dolor no esté claro, se puede optar por realizar una prueba terapéutica con la administración de zoledronato por vía intravenosa. Cuando el dolor mejora puede atribuirse a actividad de la enfermedad; cuando no hay alivio del dolor será necesario valorar completar la evaluación del paciente para identificar su causa.
En general, el tratamiento de la EOP se plantea en pacientes en los que ya existe una afectación ósea establecida, con una alteración y deformidad del hueso, con las consiguientes secuelas para el paciente. En la actualidad no existen tratamientos médicos que permitan la reversión de los cambios óseos, por lo que no está clara la utilidad del tratamiento a la hora de prevenir la progresión a osteoartrosis, revertir la deformidad ósea, mejorar la hipoacusia en el caso de la afectación del cráneo o aliviar la compresión de los nervios relacionada con la deformidad del hueso. Tampoco hay evidencia respecto a la influencia de los tratamientos sobre la calidad de vida del paciente. En todos estos casos, si no hay dolor, no hay una indicación clara de tratamiento. Está en debate si la presencia de EOP asintomática, pero con actividad bioquímica que presentan lesiones en localizaciones óseas donde existe un riesgo de que la enfermedad pueda dar lugar a complicaciones (cerca de las articulaciones, en cráneo, en columna, con presencia de focos líticos de gran tamaño) podría ser un motivo de tratamiento pese a la falta de evidencia sobre la utilidad de este. El ensayo clínico PRISM-EZ59 comparó 2 estrategias terapéuticas en la EOP: a) guiar el tratamiento con el objetivo de controlar el dolor del paciente o b) guiar el tratamiento con el objetivo de controlar la elevación de los marcadores de formación ósea (FA). La estrategia dirigida a controlar la elevación de la FA no aportó beneficio a los pacientes, ya que presentó una tendencia al aumento en el número de fracturas y de intervenciones de cirugía traumatológica sin mejorar ni el control del dolor ni la calidad de vida de los pacientes. El objetivo del tratamiento de los pacientes con EOP debe ser el control de los síntomas, sin obsesionarse por conseguir una supresión de los marcadores bioquímicos de recambio óseo. No existe por tanto un intervalo establecido de administración para los bisfosfonatos, ya que para muchos pacientes una sola dosis será suficiente, mientras que otros necesitarán repetir la dosis meses o años después de la inicial.
Además de los tratamientos farmacológicos, en los pacientes con EOP puede ser necesario valorar tratamiento de cirugía ortopédica y traumatológica dada la presencia de fracturas, deformidades óseas, osteoartrosis o compresiones nerviosas. No existe contraindicación en estos pacientes para la realización de cirugía traumatológica como las osteotomías, la cirugía para la fijación de fracturas, las artroplastias o la cirugía de columna para la descompresión de los nervios. La deformidad ósea y el aumento de vascularización del hueso afecto supondrán un reto adicional para los traumatólogos dadas las dificultades técnicas que pueden generar. Los resultados de distintas series señalan un buen resultado en las artroplastias y una buena consolidación en el hueso pagético con la excepción del fémur proximal. El uso de zoledronato por vía intravenosa previo a la cirugía podría reducir el riesgo de sangrado durante esta, aunque no existe evidencia de esta indicación. En el caso del uso prequirúrgico de bisfosfonato, la mayor preocupación es la posible repercusión que podría tener la acción del fármaco sobre la consolidación de las fracturas. Tampoco existe contraindicación para el uso de implantes cocleares o audífonos en pacientes con hipoacusia.
En las guías clínicas de las sociedades europea y americana (European Calcified Tissues Society y American Society of Bone and Mineral Research) se establecen una serie de recomendaciones para el tratamiento de la EOP60. En la figura 5 se resumen algunas de las principales.
Pronóstico
El pronóstico de la EOP es variable. Muchos de los pacientes permanecen asintomáticos. En la mayoría de los pacientes con dolor, una única administración de zoledronato a lo largo de su vida consigue una mejoría del dolor y un control de las cifras de FA. En próximos años, los resultados del estudio ZiPP46 permitirán conocer si la administración de zoledronato en individuos sanos portadores de mutaciones en el SQSTM1 puede alterar el pronóstico de la enfermedad evitándola o retrasando su inicio.
FinanciaciónNo se ha recibido financiación para la realización del artículo.
Conflicto de interesesNo se declara ningún conflicto de interés.
Quiero agradecer al Dr. del Pino Montes, internista y reumatólogo recientemente jubilado, toda su dedicación a la EOP. El Dr. del Pino es una figura pionera en el estudio de esta enfermedad en España. Su aportación fue clave para el conocimiento y la caracterización del foco de alta prevalencia de Vitigudino, en la provincia de Salamanca, uno de los focos de alta prevalencia de la enfermedad mejor conocidos en el mundo. Sin su pasión por esta enfermedad, esta revisión no hubiera podido realizarse.
