



Los tumores secretores de catecolaminas derivan de la médula adrenal o de los ganglios del sistema nervioso autónomo, y se conocen, respectivamente, como feocromocitomas y paragangliomas. A pesar de que las manifestaciones clínicas son similares, su diferenciación es importante por la posibilidad de formar parte de un síndrome endocrino hereditario y el diferente pronóstico.
Catecholamine-secreting tumors derive from the adrenal medulla or autonomic nervous system ganglia and are known as pheochromocytomas and paragangliomas, respectively. Although the clinical manifestations are similar, their differentiation is important due to the possibility of being part of a hereditary endocrine syndrome and the different prognosis.
Los tumores secretores de catecolaminas derivan de la médula adrenal o de los ganglios del sistema nervioso autónomo y se conocen como feocromocitomas y paragangliomas, respectivamente. A pesar de que las manifestaciones clínicas son similares, su diferenciación es importante por la posibilidad de formar parte de un síndrome endocrino hereditario y el diferente pronóstico.
Presentación del casoPresentamos el caso de una paciente de 16 años, sin antecedentes de interés, que refería episodios de cefalea pulsátil, diaforesis y palidez cutánea de unos 15-30 min de duración desde hacía tres años. En los meses previos al ingreso habían aumentado de frecuencia e intensidad, y se acompañaban de dolor torácico y palpitaciones.
En el último año la paciente había acudido en varias ocasiones a su centro de salud por este motivo, detectando en alguna visita cifras elevadas de tensión arterial (TA). Ante el diagnóstico de sospecha de feocromocitoma/paraganglioma, se solicitó determinación de catecolaminas en orina de 24 h con los siguientes resultados: metanefrina (mcg/24 h) 123 (52-341), normetanefrina (mcg/24 h) 7.598 (88-444), dopamina (mcg/24 h) 717,4 (46-277), metoxitiramina (mcg/24 h) 1.041 (103-434).
Como antecedentes familiares relevantes destacaba hipertensión arterial (HTA) en su abuela materna, diagnosticada a los 60 años, y bien controlada con dos fármacos (se determinaron catecolaminas en orina de 24 h que resultaron normales).
Al ingreso, la paciente se encontraba afebril, con cifras de TA mantenidas en torno a 110/70, sin tratamiento, pero con tendencia a la taquicardia. A la exploración física presentaba un fenotipo normal. Ascultación cardíaca: tonos rítmicos, soplo sistólico 2/6 en foco aórtico. Auscultación respiratoria anodina. El abdomen era blando y depresible, sin masas ni visceromegalias a la palpación.
La elevación de metanefrinas en orina se confirmó en una segunda determinación. Se amplió el estudio bioquímico con determinación de cromogranina A, perfil tiroideo, metabolismo fosfocálcico, calcitonina y antígeno carcinoembrionario (CEA). Se realizó un ecocardiograma transtorácico que no evidenció criterios de hipertrofia ventricular ni alteraciones valvulares.
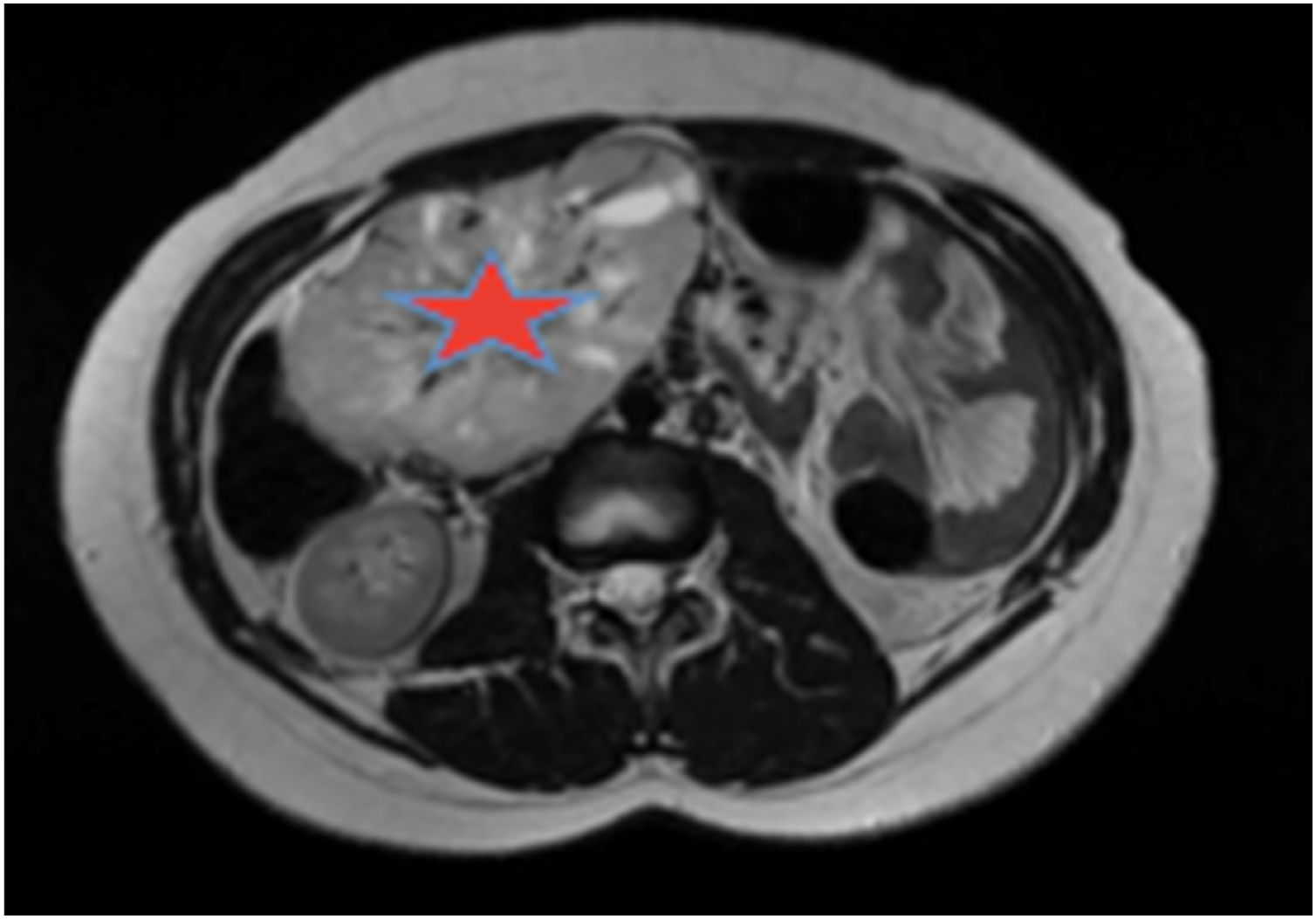
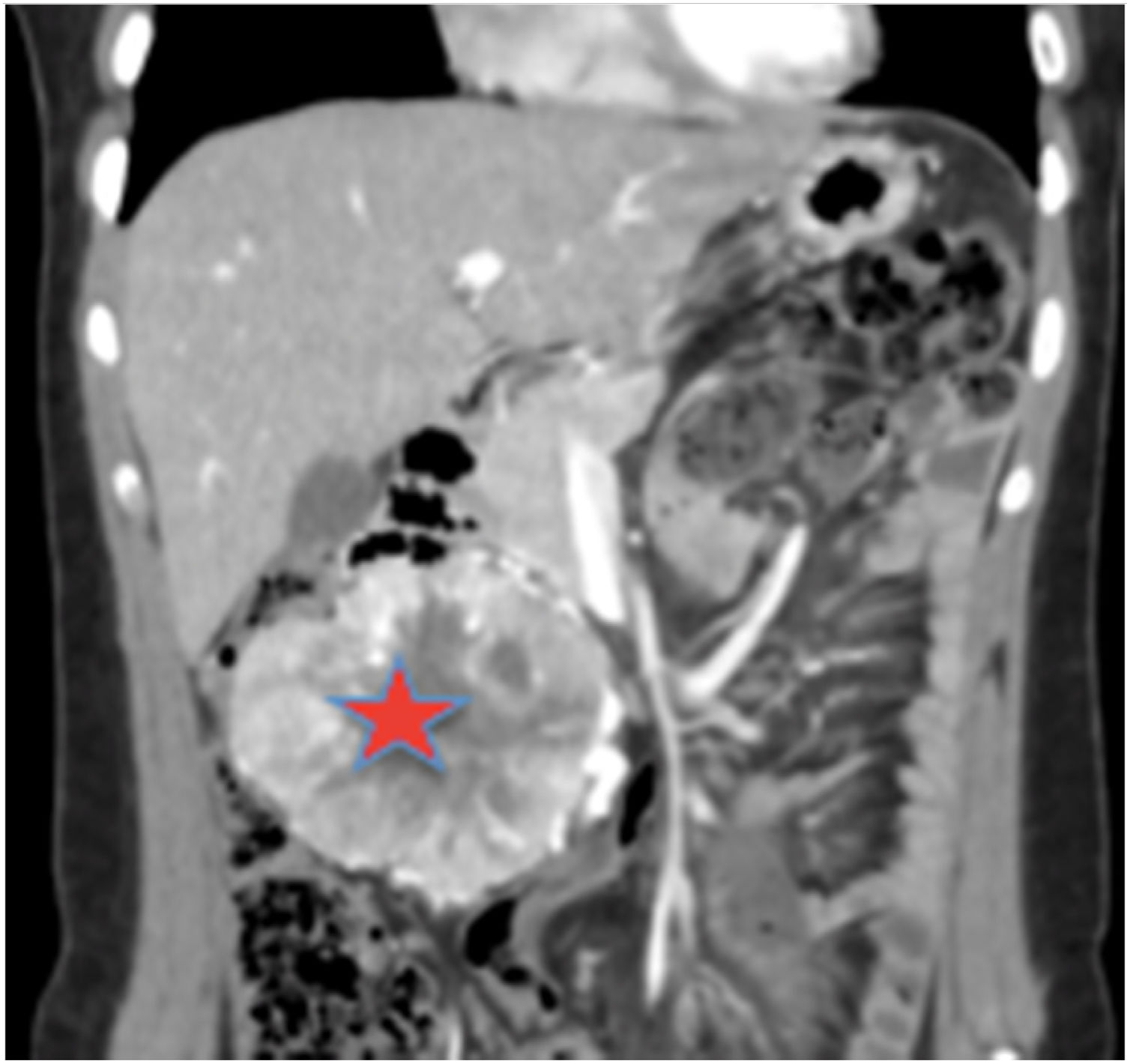
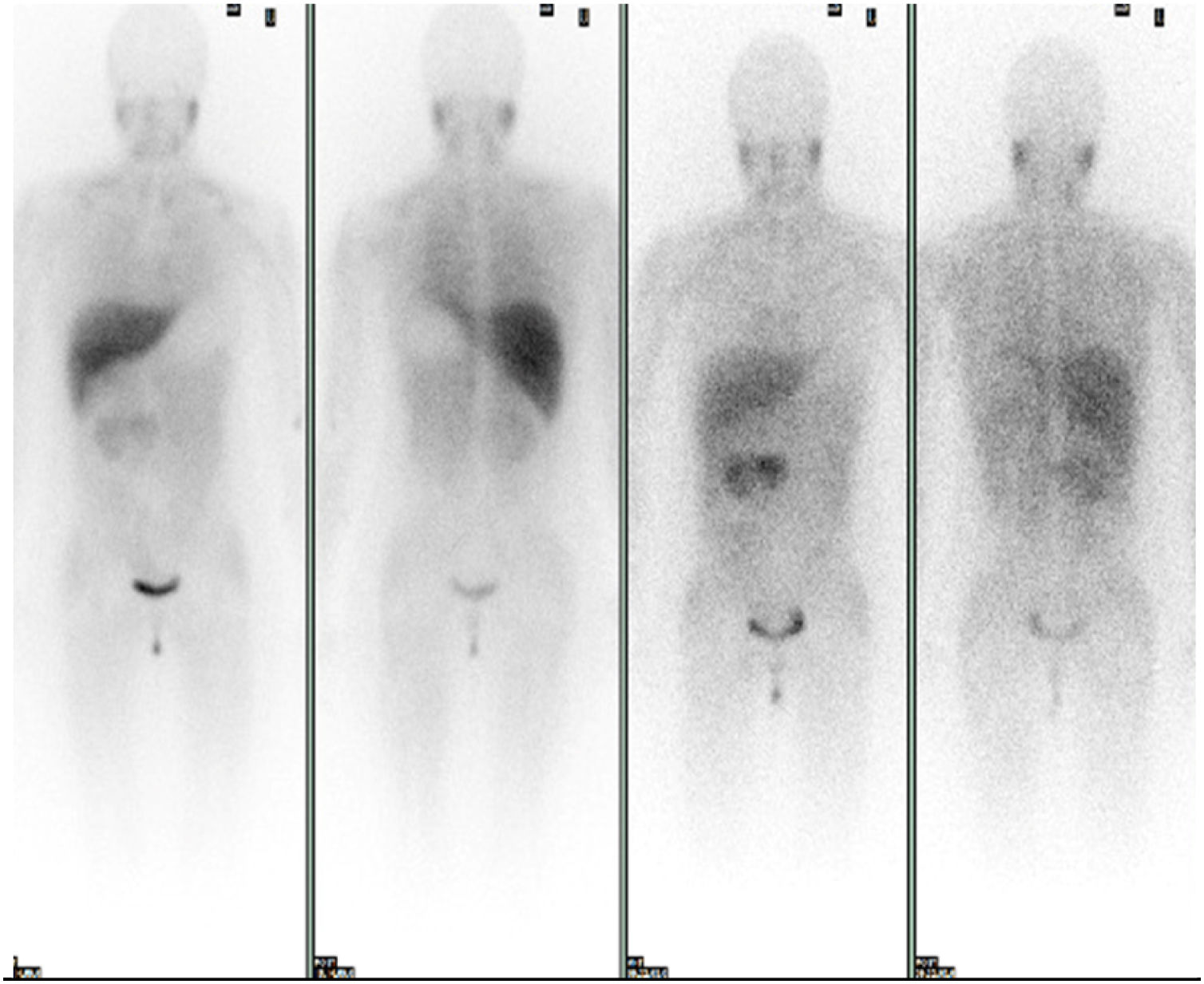
Se solicitó un estudio de localización mediante resonancia magnética nuclear (RMN) abdominal (figura 1 del anexo), que mostró una gran masa abdominal a nivel paraaórtico derecho compatible con un paraganglioma intraabdominal y estudio de extensión mediante tomografía computarizada (TC) toracoabdominal con contraste (figura 2 anexo) y gammagrafía con MIBG 131 (figura 3 anexo), que no mostraron lesiones a ningún otro nivel.

Estudio de localización: RMN abdominal. TAC T-A con contrase que evidencia gran masa de origen retroperitoneal, de 11 x 8 x 7 cm, compatible con paraganglioma paraórtico derecho. La lesión está altamente vascularizada, con vasos arteriales y venosos de gran calibre en su interior.
, que condiciona efecto masa con compresión de la vena cava inferior y desplazamiento de asas intestinales.")

Estudio gammagráfico con MIB 131. Estudio de extensión con MIBG 131. Se observa captación fisiológica en hígado y salivares y eliminación urinaria. En el área cardiaca existe una actividad muy tenue e irregular, en probable relación con el exceso de catecolaminas. Se objetiva presencia de receptores adrenérgicos en la masa abdominal conocida.
Una vez confirmado el diagnóstico, se inició tratamiento alfa-bloqueante con fenoxibenzamina a dosis progresivas (dosis final 10 mg/12 h) y a los cinco días se añadió tratamiento beta-bloqueante por taquicardia refleja (atenolol 25 mg/24 h). A los 20 días fue intervenida mediante laparotomía con extracción de masa extraadrenal derecha de 10 cm retroperitoneal (retrocava) y linfadenectomía peritumoral periaortocava e infrarrenal.
La anatomía patológica demostró un paraganglioma pobremente diferenciado con presencia de dos metástasis ganglionares y valores de Ki 67 > 3%, alcanzando en algunas áreas un 20%.
El estudio genético mostró una mutación en el gen SDHB (portadora en heterocigosis de la variante de significado desconocidoc.424-3C>G en el gen SDHB). El estudio de los progenitores ha evidenciado que el padre de la paciente es portador de la mutación.
Hasta la fecha, la paciente permanece asintomática, con buen control de TA. La determinación seriada de metanefrinas en orina de 24 h y pruebas de imagen (RMN de abdomen y tomografía por emisión de positrones-tomografía computarizada con fluorodesoxiglucosa [PET-TAC FDG]) no muestran recidiva ni metástasis.
DiscusiónEl tratamiento de los tumores secretores de catecolaminas es complejo. Los factores histológicos que determinan una mayor agresividad no están bien definidos, aunque el tamaño > 5 cm1, la localización extra-adrenal2, la mutación SDHB y FH2–8, la elevación de dopamina y de su metabolito 3 metoxitiramina8–11, así como una edad temprana al diagnóstico11 se han asociado con mayor riesgo de recurrencia y enfermedad metastásica.
En la actualidad no está establecido ningún protocolo específico para el seguimiento radiológico de los feocromocitomas y paraganglioma. La última guía de la Sociedad Europea de Endocrinología recomienda combinar en el seguimiento estudios de imagen anatómica y funcional en las formas con mayor riesgo de enfermedad metastásica, dependiendo del síndrome hereditario o localización del tumor12,13. En el caso de paragangliomas por mutación de SDHB, la RMN y la PET con 18F-FDG o con 68Ga-DOTA-Tyr3-octreótido (68Ga-DOTA-TOC) parecen tener mayor sensibilidad y especificidad14,15.
Es necesario un seguimiento a largo plazo, incluso en pacientes aparentemente curados y especialmente en aquellos con mayor riesgo de malignidad, ya que pueden presentar recurrencias o enfermedad metastásica hasta varios años después del diagnóstico12,13.
El estudio genético de esos tumores es clave para conocer el pronóstico, adecuar el tratamiento e individualizar el seguimiento8,12–15.
FinanciaciónNo se ha recibido financiación por parte de ninguna entidad.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
