






La distrofia muscular miotónica tipo 1 (DM1) o enfermedad de Steinert (CIE-9-C: 359.21; CIE-10-ES: G71.11, ORPHA: 273) es una miopatía autosómica dominante de baja prevalencia (<5/10.000) con penetrancia casi completa y daño multiorgánico (neurológico, cardiológico, respiratorio, endocrinológico y digestivo). Es una de las enfermedades humanas con mayor variabilidad clínica. Los síntomas más incapacitantes o molestos para estos enfermos (limitación de la movilidad, cansancio crónico, somnolencia diurna o trastornos digestivos) y sus familias (apatía y falta de iniciativa) no son necesariamente los de peor pronóstico. Las complicaciones respiratorias y los trastornos cardíacos reducen la esperanza de vida de los afectados. No existe tratamiento que modifique su evolución. La función del médico de atención primaria es decisiva en el seguimiento de la DM1, ya sea coordinando a las diferentes especialidades implicadas en el mismo o detectando las complicaciones tratables, en las cuales se centra el presente trabajo.
Myotonic dystrophy type 1 (DM1) or Steinert's disease (CIE-9-C: 359.21; CIE-10-ES: G71.11, ORPHA: 273) is a rare autosomal dominant inherited myopathy with almost complete penetrance and multisystemic consequences (neurological, cardiological, respiratory, endocrinological, and gastrointestinal). It is one of the clinical most variable diseases. The most bothersome symptoms for the patients (mobility problems, fatigue, hypersomnia, or gastrointestinal symptoms) and their families (apathy, lack of initiative) are not necessarily the most dangerous. Respiratory problems and cardiac arrhythmias shorten life expectancy. There is no specific treatment. The role of the Primary Care physician is crucial in the follow-up of DM1, either by coordinating the different professionals or detecting treatable complications. This work addresses the latter.
La enfermedad de Steinert o distrofia miotónica tipo 1 (DM1) es una miopatía caracterizada por miotonía, atrofia y debilidad muscular con afectación multiorgánica, cuyas características clínicas más destacables son los problemas respiratorios, arritmias por defectos en el sistema de conducción del músculo cardíaco, cataratas precoces, hipogonadismo, resistencia a la insulina, hipersomnia y calvicie frontal. Es la más frecuente de las distrofias musculares de aparición en la edad adulta (en promedio, un caso cada 8.000 personas). Se asocia con anomalías en el locus 19q13.3 (repetición anormalmente elevada del triplete citosina, timina, guanina [CTG]). De transmisión autosómica dominante, puede presentar el fenómeno de anticipación, consistente en que la enfermedad se agrava y aparece en la descendencia a una edad más temprana en sucesivas generaciones1. La detección de la anomalía genética utilizando técnicas moleculares confirma el diagnóstico. No obstante, el asesoramiento genético no resulta fácil debido a la amplia variabilidad en la expresión clínica, tanto intrafamiliar como entre distintas familias. Se aconseja el diagnóstico prenatal sobre todo para la transmisión materna debido a la gravedad de las formas neonatales2. Carece de tratamiento que modifique su evolución. Su atención médica debería ser tanto multidisciplinar, con la participación de varias especialidades clínicas, como multiprofesional, con intervención de otras profesiones sanitarias no médicas. Sin embargo, la asistencia médica de la enfermedad de Steinert ha sido tradicionalmente fragmentaria, insuficiente o inexistente3.
Se ha descrito una miopatía miotónica proximal o distrofia miotónica tipo 2 (DM2), menos prevalente que la DM1 y de características más benignas4.
La evolución de la enfermedad suele ser lenta y progresiva, aunque a veces se produce un deterioro rápido. La esperanza de vida de estos enfermos es varios lustros inferior a la de la población general, debido sobre todo a complicaciones respiratorias5.
Para facilitar su seguimiento integrado, se ha elaborado una guía clínica de la DM1, a la que se aludirá a lo largo del presente trabajo6.
EpidemiologíaAunque la prevalencia mundial estimada de la DM1 es de 12,5 casos por cada 100.000 habitantes7, existen variaciones regionales. En la Comunidad de Madrid, se ha encontrado una prevalencia de 13,3/100.0008.
EtiopatogeniaEl defecto genético consiste en una expansión del trinucleótido CTG localizado en la región 3’ no codificante del gen de la proteína cinasa de la distrofia miotónica (Dystrophia Myotonica Protein Kinase [DMPK]) ubicada en el brazo largo del cromosoma 19 (19q13.3). La transcripción de estas repeticiones resulta en una expansión y acumulación de inclusiones ribonucleares en el núcleo, con el resultado final de una alteración del corte y empalme de múltiples genes como los que codifican el canal de cloro tipo 1 (CLC-1), el receptor de la insulina y el de la troponina T cardíaca, entre otros9.
La clínica y progresión de la DM1 dependen de la cantidad de repeticiones CTG: no suele haber signos clínicos en individuos con 50-100 repeticiones de CTG, pero existe correlación entre el tamaño de la repetición y la edad de comienzo de la DM1 cuando el número de repeticiones es <400. El fenómeno de anticipación obedece a la acumulación de tripletes CTG en sucesivas generaciones. Aunque las repeticiones patológicas del triplete CTG se producen en todos los tejidos, existe entre ellos una gran heterogeneidad. Así, el tamaño de la expansión es mayor en el músculo que en leucocitos de sangre periférica (utilizados en el diagnóstico molecular). Además, el tamaño de la expansión puede incrementarse a lo largo de la vida del sujeto (mutación dinámica). Aunque la base genética de la DM1 ha quedado establecida, los mecanismos moleculares por los que la expansión del triplete CTG da lugar al fenotipo de la DM1 todavía no son totalmente conocidos4.
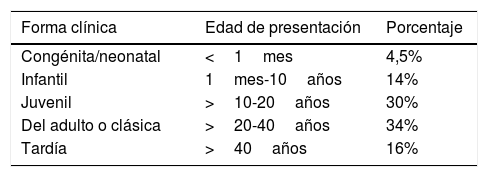
Clínica y diagnósticoPuede manifestarse en cualquier etapa de la vida, con 5 formas de presentación en relación con el número de repeticiones (tabla 1)10. En adolescentes y adultos la primera manifestación suele ser la miotonía (contracción muscular con dificultad para la relajación) que limita o impide soltar objetos asidos con la mano. A veces pasa desapercibida, empeora con el frío y mejora al calentar el músculo. La debilidad y atrofia musculares se instauran progresivamente en los músculos faciales, masticatorios, del cuello, flexores de los dedos, antebrazos y flexores de los pies. La debilidad de las extremidades es de predominio distal e intensidad variable, con hiperextensión en reposo de los dedos de la mano por atrofia de la musculatura flexora. Esta debilidad distal de extremidades superiores es referida por los pacientes como dificultad para abrir latas o botes y en las extremidades inferiores se manifiesta en forma de tropiezos y caídas frecuentes.
Formas clínicas o fenotipos de presentación de la DM1
| Forma clínica | Edad de presentación | Porcentaje |
|---|---|---|
| Congénita/neonatal | <1mes | 4,5% |
| Infantil | 1mes-10años | 14% |
| Juvenil | >10-20años | 30% |
| Del adulto o clásica | >20-40años | 34% |
| Tardía | >40años | 16% |
Fuente: Illa et al.10.
El fenotipo facial de estos pacientes resulta característico: cara alargada y estrecha con sienes hundidas, ptosis palpebral, boca entreabierta en reposo, voz gangosa, cuello curvado hacia delante y calvicie frontal (fig. 1)11.
. Madrid, Museo Nacional del Prado.")
La función del médico de atención primaria (MAP) en la DM1, desde el punto de vista diagnóstico, consiste en sospechar la existencia de una miopatía12. La evaluación del paciente con debilidad muscular requiere una anamnesis y exploración física cuidadosas, a fin de diferenciar la debilidad muscular de otros síntomas (astenia, fatiga, dolor, rigidez o pérdida de fuerza de origen no muscular). La distribución de la debilidad muscular (generalizada, localizada asimétrica o localizada simétrica, proximal o distal) puede orientar sobre su etiología y contribuir a diferenciar trastornos neurológicos y estrictamente miopáticos.
El enfermo con miopatía puede presentar una elevación aislada de la creatina-(fosfo)-cinasa (CPK) aislada, sin otra clínica acompañante. Cualquier proceso con lesión muscular (traumatismos, quemaduras, cirugías, inyecciones intramusculares, biopsias, decúbito prolongado o ejercicio intenso) puede elevar el nivel de CPK. Pero únicamente las elevaciones por encima de 5 veces su valor normal, es decir, superiores a 1.000UI/l, indican claramente la existencia de miopatía. Aunque algunas cursan con niveles de CPK menores, cifras inferiores a 1.000UI/l y, especialmente, por debajo de 500UI/l son inespecíficas. Como las determinaciones de GOT y GPT se realizan de forma rutinaria, pueden encontrarse hipertransaminasemias asintomáticas que, sobre todo en un paciente joven, se deben complementar con una determinación de CPK que permite saber fácilmente si esta elevación de GOT y GPT es de origen muscular o hepático. Ante la sospecha de miopatía, conviene precisar con exactitud el tipo de miopatía de que se trata. Prácticamente todas las miopatías pueden presentarse como una elevación de CPK asintomática y, en muchos casos, este trastorno analítico será la única manifestación de la enfermedad. Como la mayoría de las miopatías tienen una base genética, el paciente puede transmitir la enfermedad a su descendencia y el fenotipo de los hijos puede no ser el mismo que el del padre o la madre. El asesoramiento genético solo es practicable con un diagnóstico de la miopatía lo más exacto posible13.
El diagnóstico de confirmación de la DM1 es competencia del neurólogo y se basa, junto con la exploración clínica, en el estudio electrofisiológico (electromiografía [EMG]) y en el estudio genético. Dada la especificidad del diagnóstico molecular, actualmente no es necesario realizar biopsia muscular a un paciente con sospecha de DM114.
Afectación neurológicaLa aplicación de las diferentes escalas para la valoración funcional de la DM1y el eventual tratamiento farmacológico de la miotonía se circunscriben al ámbito de la atención especializada (tabla 2).
- •
Caídas. Los enfermos con DM1 sufren caídas 10 veces más que la población general. Las fracturas más frecuentes descritas fueron del tobillo y del pie, casi la mitad del total, pero no se informó de fracturas de cadera15. Las causas son múltiples: debilidad permanente y progresiva de grupos musculares críticos para la marcha (extensores de la rodilla y flexores dorsales del pie) que alteran el equilibrio al caminar, déficits visuales (ptosis palpebral, cataratas), deterioro muscular por inactividad y alteraciones cognitivas. La trascendencia clínica de las fracturas en las extremidades distales en las miopatías progresivas radica en que pueden dar lugar a una pérdida funcional permanente y a la dependencia de una silla de ruedas.
- •
Hipersomnia diurna. Muy frecuente en la DM1, con patrón similar al de la narcolepsia. En una minoría de enfermos, es incapacitante para la vida laboral y social. Aunque es habitual la presencia de un síndrome de apnea/hipopnea del sueño, también parece existir un trastorno de la regulación de origen central16.
- •
Deterioro cognitivo. En conjunto, los rasgos de personalidad y síntomas psicológicos de los pacientes con DM1 se sitúan en el intervalo medio de la normalidad en cuanto a niveles de autoestima e ideación suicida. No obstante, se ha encontrado que un 27% presenta un alto riesgo de desarrollar un trastorno psiquiátrico. De hecho, los rasgos psicológicos difieren entre los distintos fenotipos, con síntomas más graves en los fenotipos más graves. La presencia de una mayor ansiedad fóbica y de una menor autoestima se asociaron con un menor nivel educativo, un mayor número de repeticiones CTG, una afectación muscular más grave y un menor funcionamiento cognitivo17.
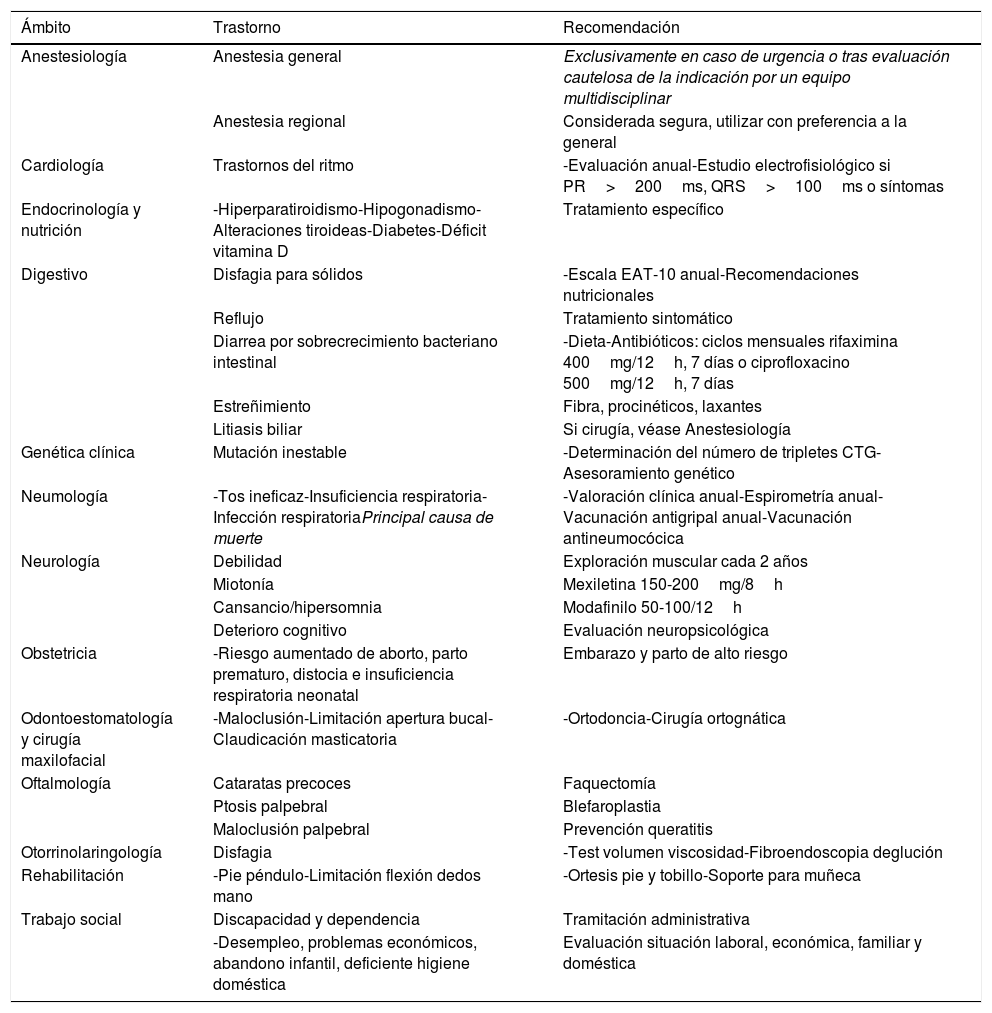
Recomendaciones para el seguimiento de la DM1
| Ámbito | Trastorno | Recomendación |
|---|---|---|
| Anestesiología | Anestesia general | Exclusivamente en caso de urgencia o tras evaluación cautelosa de la indicación por un equipo multidisciplinar |
| Anestesia regional | Considerada segura, utilizar con preferencia a la general | |
| Cardiología | Trastornos del ritmo | -Evaluación anual-Estudio electrofisiológico si PR>200ms, QRS>100ms o síntomas |
| Endocrinología y nutrición | -Hiperparatiroidismo-Hipogonadismo-Alteraciones tiroideas-Diabetes-Déficit vitamina D | Tratamiento específico |
| Digestivo | Disfagia para sólidos | -Escala EAT-10 anual-Recomendaciones nutricionales |
| Reflujo | Tratamiento sintomático | |
| Diarrea por sobrecrecimiento bacteriano intestinal | -Dieta-Antibióticos: ciclos mensuales rifaximina 400mg/12h, 7 días o ciprofloxacino 500mg/12h, 7 días | |
| Estreñimiento | Fibra, procinéticos, laxantes | |
| Litiasis biliar | Si cirugía, véase Anestesiología | |
| Genética clínica | Mutación inestable | -Determinación del número de tripletes CTG-Asesoramiento genético |
| Neumología | -Tos ineficaz-Insuficiencia respiratoria-Infección respiratoriaPrincipal causa de muerte | -Valoración clínica anual-Espirometría anual-Vacunación antigripal anual-Vacunación antineumocócica |
| Neurología | Debilidad | Exploración muscular cada 2 años |
| Miotonía | Mexiletina 150-200mg/8h | |
| Cansancio/hipersomnia | Modafinilo 50-100/12h | |
| Deterioro cognitivo | Evaluación neuropsicológica | |
| Obstetricia | -Riesgo aumentado de aborto, parto prematuro, distocia e insuficiencia respiratoria neonatal | Embarazo y parto de alto riesgo |
| Odontoestomatología y cirugía maxilofacial | -Maloclusión-Limitación apertura bucal-Claudicación masticatoria | -Ortodoncia-Cirugía ortognática |
| Oftalmología | Cataratas precoces | Faquectomía |
| Ptosis palpebral | Blefaroplastia | |
| Maloclusión palpebral | Prevención queratitis | |
| Otorrinolaringología | Disfagia | -Test volumen viscosidad-Fibroendoscopia deglución |
| Rehabilitación | -Pie péndulo-Limitación flexión dedos mano | -Ortesis pie y tobillo-Soporte para muñeca |
| Trabajo social | Discapacidad y dependencia | Tramitación administrativa |
| -Desempleo, problemas económicos, abandono infantil, deficiente higiene doméstica | Evaluación situación laboral, económica, familiar y doméstica |
Fuente: Adaptado de Gutiérrez et al.6.
Se ha tendido a explicar la conducta apática de estos enfermos basándose en las alteraciones musculares, psicológicas o psiquiátricas asociadas a la DM1. Sin embargo, la disfunción cognitiva ha recibido menos atención, aunque puede afectar seriamente a la calidad de vida18. La comparación entre un grupo de pacientes con DM1, un grupo de afectados por distrofia facioescapulohumeral (DFSH) con un grado de discapacidad similar, y un grupo control, demostró que la apatía es independiente del ámbito psicopatológico, del cansancio, de la edad y de la discapacidad motora, pero que se asocia en general con el estado cognitivo. En conjunto, estos resultados sugieren que la apatía es de origen central y no el resultado del proceso de afrontamiento de una enfermedad invalidante19. Por otra parte, la comparación de la capacidad intelectual entre las diferentes formas clínicas de la DM1 tardías ha puesto de manifiesto que el deterioro intelectual es una característica importante y muy frecuente de esta enfermedad, no solo en los fenotipos o formas clínicas con coeficientes intelectuales inferiores a los de la población general, sino también en la forma del adulto o clásica20.
CardiopatíasAunque la mayoría de los enfermos carece de síntomas, es frecuente que el corazón se vea afectado. Puede estarlo incluso en pacientes asintomáticos desde el punto de vista musculoesquelético, dado que no existe correlación entre la gravedad de la afectación muscular y las manifestaciones cardíacas. Los trastornos más frecuentes son las alteraciones del sistema de conducción de His-Purkinje. Se han descrito defectos de conducción auriculoventricular e intraventricular, bloqueo de rama derecha, bloqueo fascicular anterior izquierdo y bloqueo auriculoventricular. Se ha comunicado la aparición de bradicardia sinusal, extrasístoles auriculares y ventriculares, fibrilación, flúter auricular y taquicardia ventricular. Pese a su alta incidencia, casi todas las alteraciones de la conducción y del ritmo en estos enfermos son leves y, por tanto, subclínicas.
En pacientes con DM1 seguidos de forma protocolizada en una consulta monográfica, se identificó bradicardia sinusal en el 48,8%, disfunción sinusal en el 13,8%, arritmias supraventriculares en el 10%, intervalo PR>220ms en el 31,3%, taquicardia ventricular en el 5%, intervalo QT corregido largo en el 5%, bloqueo auriculoventricular de segundo o tercer grado en el 8,8% e intervalo QRS>120ms en el 7,5%. Únicamente un paciente presentó disfunción ventricular grave. Durante el seguimiento se implantaron 15 marcapasos y 2 desfibriladores y se realizaron 5 estudios electrofisiológicos, la mayoría de ellos por taquicardia ventricular. Solo se produjo una muerte súbita21.
La falta de correlación entre la intensidad de la afectación muscular y las alteraciones cardíacas exige que el MAP complemente las recomendaciones de la tabla 2 como sigue:
- •
Informando al paciente sobre síntomas de alarma (síncope, palpitaciones) que justifican una consulta urgente.
- •
Realizando una anamnesis dirigida anual, interrogando específicamente sobre la presentación de dichos síntomas de alarma.
- •
Pautando un ECG anual para valorar la conveniencia de un estudio electrofisiológico.
Los problemas respiratorios son frecuentes y la principal causa de muerte prematura en estos pacientes. Aunque se suelen diagnosticar con retraso, debido a su instauración insidiosa en pacientes que ya presentan manifestaciones musculares y sistémicas, a veces la insuficiencia respiratoria aparece de forma aguda desencadenada por un proceso anestésico o una infección respiratoria.
Las infecciones respiratorias recurrentes, la disnea progresiva y la hipersomnia diurna con cefalea matutina deben hacer pensar al MAP en la existencia de una afectación respiratoria hasta entonces desapercibida.
Debido al carácter complejo de las alteraciones respiratorias de la DM1, que pueden coexistir con diferentes grados de trastornos del sueño y cognitivos, se ha elaborado un consenso para el abordaje de la insuficiencia respiratoria crónica en esta enfermedad que recoge recomendaciones para identificar la existencia de afectación respiratoria (tabla 3)22. La primera evaluación de la función pulmonar no es necesario que sea realizada por el neumólogo. Puede y debe hacerla el médico responsable del seguimiento del paciente, ya sea el MAP o el neurólogo. Este interrogatorio deberá preceder a las pruebas complementarias respiratorias recomendadas en la tabla 2.
Orientaciones para identificar la insuficiencia respiratoria en la DM1
| 1.- Ortopnea |
| ¿Le falta aliento estando acostado? |
| ¿Necesita dormir con más de una almohada? |
| ¿Se duerme sentado en una silla o sillón? |
| 2.- Disnea en las actividades de la vida diaria |
| ¿Le falta aliento al moverse por la casa? |
| ¿Le falta aliento al lavarse o vestirse? |
| ¿Le falta aliento al caminar? |
| 3.- Sueño de mala calidad |
| ¿Se siente cansado al levantarse por la mañana? |
| ¿Se despierta más de una vez por la noche, además de para ir al baño? |
| ¿Necesita levantarse de la cama por piernas inquietas? |
| 4.- Cefalea matutina |
| ¿Se despierta por la mañana con dolor de cabeza? |
| ¿Siente la cabeza pesada por la mañana? |
| ¿Siente presión en la cabeza al despertar? |
| 5.- Apneas |
| ¿Le falta aliento durante la noche? |
| ¿Le han dicho que hace pausas en la respiración durante el sueño? |
| ¿Le han dicho que deja de roncar y empieza de nuevo de forma súbita durante el sueño? |
| 6.- Deterioro cognitivo |
| ¿Percibe que su capacidad de concentración ha empeorado? |
| ¿Nota que piensa más despacio de lo habitual? |
| ¿Tiene menos motivación de la habitual para realizar actividades? |
| 7.- Somnolencia diurna excesiva |
| ¿Se duerme mientras come? |
| ¿Tiende a dormirse conduciendo? |
| ¿Se duerme mientras le hablan? |
| 8.- Cansancio |
| ¿Se siente más cansado de lo habitual? |
| ¿Se siente cansado al despertar tras el sueño nocturno? |
| ¿Se siente más cansado de lo esperado tras las actividades del día? |
| 9.- Tratamiento de infecciones respiratorias desde la última visita |
| ¿Ha tenido alguna infección respiratoria que requiera tratamiento? |
| ¿Ha utilizado antibióticos por tos intensa? |
| ¿Ha ingresado en el hospital por infección respiratoria o falta de aliento? |
Fuente: Adaptado de22.
Los autores del consenso para el cribado respiratorio advierten que los pacientes con DM1 necesitan un tiempo adicional para su entrevista y evaluación, posiblemente debido a disfunciones cognitivas y ejecutivas frecuentes incluso en enfermos con una discapacidad motora poco acusada. Los médicos deben ser conscientes de las diferencias entre estos pacientes y los afectados por otras enfermedades neuromusculares.
Se debe resaltar una vez más la importancia de identificar limitaciones respiratorias poco sintomáticas, que pueden manifestarse de forma inesperada tras procedimientos anestésicos o procesos infecciosos banales, pues, como quedó dicho, las complicaciones respiratorias son la primera causa de muerte de estos pacientes.
Endocrinología y metabolismoSe han identificado diferentes trastornos endocrinos en estos pacientes. Los más frecuentes son hipogonadismo hipergonadotropo, alteraciones tiroideas y trastornos del metabolismo hidrocarbonado y fosfocálcico. En una muestra amplia de enfermos con DM1 se encontró hiperparatiroidismo con elevación de PTH en un 80% de los casos. De estos, el 16% presentó normocalcemia, el 2% tenía hipercalcemia y un 3% adicional mostró hipercalcemia sin elevación de PTH; el 7% tuvo niveles alterados de TSH (2% por debajo de los valores normales y 5% cifras superiores a la normalidad); el 5% de los pacientes cumplió criterios de diabetes mellitus; el 17% de los varones tenían una LH aumentada y niveles plasmáticos bajos de testosterona, que indicaban la existencia de insuficiencia androgénica absoluta. Otro 21% mostró niveles elevados de LH, pero con cifras normales de testosterona, indicativas de insuficiencia androgénica relativa23.
Como la vitamina D influye en el metabolismo muscular, es necesario identificar y corregir su déficit en la DM1 para prevenir la osteomalacia (recuérdense las caídas de repetición propias de la enfermedad, con riesgo de fractura, así como el hiperparatiroidismo secundario, que empeoraría la debilidad muscular)24.
Si resultase necesario tratamiento farmacológico para la dislipidemia, es posible utilizar estatinas y fibratos, pero siempre bajo un estricto control, ya que pueden empeorar la miopatía.
La disfagia característica de la DM1, descrita seguidamente, puede favorecer la desnutrición y la pérdida de peso.
GastroenterologíaLa disfagia puede favorecer la aparición de neumonías aspirativas de gran importancia en una enfermedad cuya primera causa de mortalidad son las infecciones respiratorias. La debilidad y la miotonía de los músculos masticatorios y de la faringe reducen la capacidad de propulsión faríngea. Por último, la debilidad de la musculatura respiratoria impide producir una tos potente capaz de expulsar cuerpos extraños de las vías respiratorias.
Además de identificar y valorar la disfagia (anexo, material adicional)25, se deben facilitar a los pacientes normas nutricionales para tratarla6.
En la DM1 pueden existir alteraciones en cualquier parte del tubo digestivo, cuyo tratamiento se describe en la tabla 2.
OftalmologíaLa aparición de cataratas iridiscentes con aspecto característico («en árbol de Navidad») prematuras, por debajo de los 40 años de edad, es frecuente en la DM1. La debilidad de los músculos orbiculares facilita la aparición de ptosis palpebral, que puede llegar a la ceguera funcional por imposibilidad de abrir los ojos. Al mismo tiempo, la debilidad de la musculatura del párpado puede causar maloclusión ocular con riesgo de queratitis, que el MAP debe sospechar ante la aparición de clínica indicativa de afectación del polo anterior.
AnestesiaNo está contraindicada la utilización de anestésicos locales, salvo por motivos específicos.
El uso de anestesia regional incluyendo técnicas neuroaxiales (caudal, epidural y subaracnoidea) ha sido frecuente y se ha comunicado que es segura. La adición de opioides o tranquilizantes con efectos respiratorios debe hacerse con cautela.
La anestesia general debe emplearse exclusivamente en casos de urgencia y tras una evaluación cautelosa de la indicación por un equipo interdisciplinar. Los agentes hipnóticos intravenosos como propofol, combinado con opioides de acción corta (p.ej., remifentanilo), han sido usados con seguridad en pacientes con DM1.
En estadios precoces de la enfermedad la sedación puede realizarse con seguridad. La sedación en pacientes en estadios avanzados (afectación cardiopulmonar, debilidad muscular proximal) podría llevarse a cabo tras una evaluación del riesgo individual de fallo respiratorio y aspiración26.
Aspectos socioeconómicosSon frecuentes las carencias sociales, laborales y económicas en los pacientes con DM1. En un estudio comparativo sobre la situación de empleo en diferentes enfermedades neuromusculares, aunque se encontró que el porcentaje de pacientes con DM1 y educación superior era bajo, aquellos que la tenían presentaban una probabilidad de conseguir empleo casi 10 veces mayor que los afectados por DM1 con menor nivel educativo27. Los autores explican esta diferencia suponiendo que en los pacientes con DM1 se potenciaría desde edades tempranas la importancia de la educación, alentándoles a estudiar cuando sus condiciones lo permiten.
Junto a las limitaciones motoras propias de la DM1, existen otros factores que también pueden obstaculizar la inserción laboral de estos enfermos.
La gran variabilidad de situaciones psicológicas y cognitivas que pueden presentar los pacientes con distrofia miotónica de Steinert genera un espectro igualmente amplio de situaciones académicas, laborales y económicas, que oscilan entre la exclusión social y una perfecta integración sociolaboral.
Ante todo enfermo con DM1, el médico informado debe prescindir de ideas preconcebidas e intentar analizar cada caso de forma individualizada. En definitiva, el MAP tiene la oportunidad de conocer directamente el contexto familiar y puede detectar situaciones de carencia socioeconómica, habituales en esta enfermedad, para ponerlas en manos de los profesionales del trabajo social sanitario.
Conclusiones - puntos clave- •
La distrofia miotónica tipo 1 (DM1) o enfermedad de Steinert es una miopatía con afectación multiorgánica.
- •
Las infecciones respiratorias son la primera causa de mortalidad en la DM1.
- •
Disfagia y debilidad de la musculatura respiratoria son los principales factores predisponentes para desarrollar infecciones respiratorias en la DM1.
- •
Los afectados por la DM1 sufren 10 veces más caídas que la población general, cuyas secuelas pueden agravar la discapacidad propia de la enfermedad.
Este artículo no ha recibido financiación para su elaboración.
Conflicto de interesesNinguno.
