




La enfermedad de Behçet es una enfermedad inflamatoria multisistémica crónica que evoluciona por brotes. Es más común en Asia y en los países de la cuenca mediterránea oriental (Ruta de la Seda). En España la prevalencia es de 5 a 10 casos por 100.000 habitantes. Es una enfermedad de difícil diagnóstico por las numerosas y variadas manifestaciones clínicas y porque no se dispone de pruebas de laboratorio patognomónicas. El retraso en el diagnóstico, frecuente en países de baja prevalencia como España, aumenta la morbilidad y la mortalidad de los pacientes con enfermedad de Behçet.
Behçet's disease is an inflammatory multisystemic chronic disease that progresses by outbreaks. It is more common in Asia and countries in the eastern Mediterranean basin (Silk Route). In Spain the prevalence is between 5 and 10 cases per 100,000 inhabitants. It is a difficult disease to diagnose because of the multiple and varied clinical manifestations, and because there are not pathognomonic laboratory tests available. The delay in the diagnosis, which is frequent in countries of low prevalence like Spain, increases the morbidity and the mortality of patients with Behçet¿s disease.
El primero de los casos (caso A) corresponde a una paciente de 45 años con antecedentes de intervención por varices en los miembros inferiores en el año 2003 y de aftas orales recidivantes.
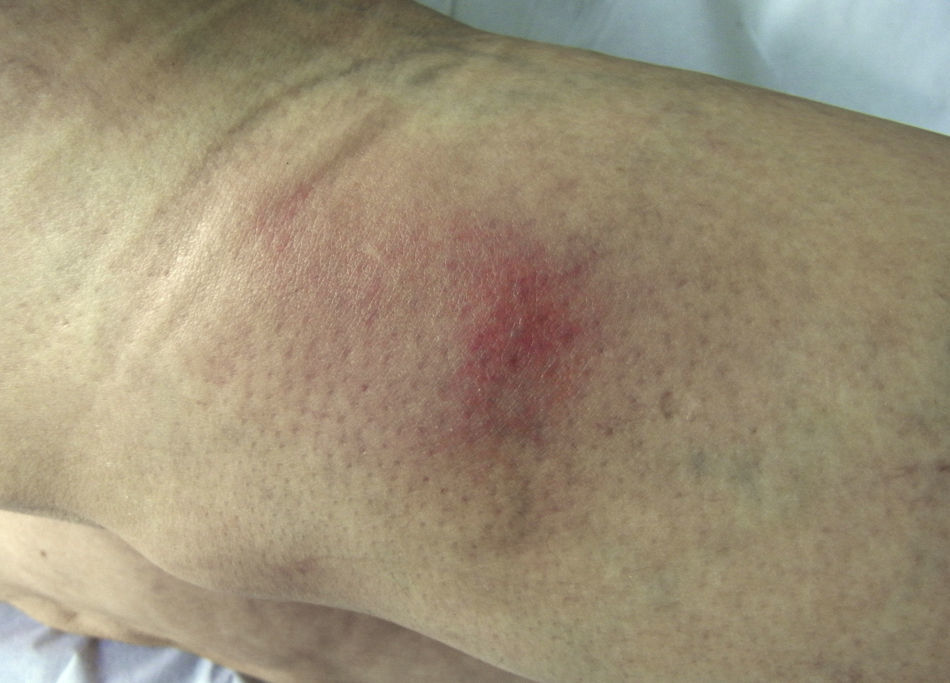
Acude a la consulta por lesiones nodulares eritematosas, calientes y dolorosas en la cara posterior de la pierna izquierda (fig. 1) y en la cara anterior de la pierna derecha. La paciente no refiere un proceso faríngeo previo ni toma anticonceptivos orales ni otros fármacos.

El diagnóstico inicial es de eritema nudoso (paniculitis) por lo que se solicita un análisis con ASLO, Mantoux y radiografía de tórax teniendo en cuenta que las causas más frecuentes de la paniculitis son la infección por estreptococo betahemolítico y la tuberculosis. Se prescribe indometacina 25 mg 1/8h, y omeprazol 20 mg 1 c/día.
La analítica de la paciente presenta: Hb 10,4g/dl, Hto 31,0, GR 3,81 millones/mm3, VCM 81,3, HCM 34,4, VSG 24, ASLO 200, glucosa, creatinina, GOT, GPT, GGT y FA normales. Mantoux de 15mm y radiografía de tórax sin alteraciones pleuropulmonares.
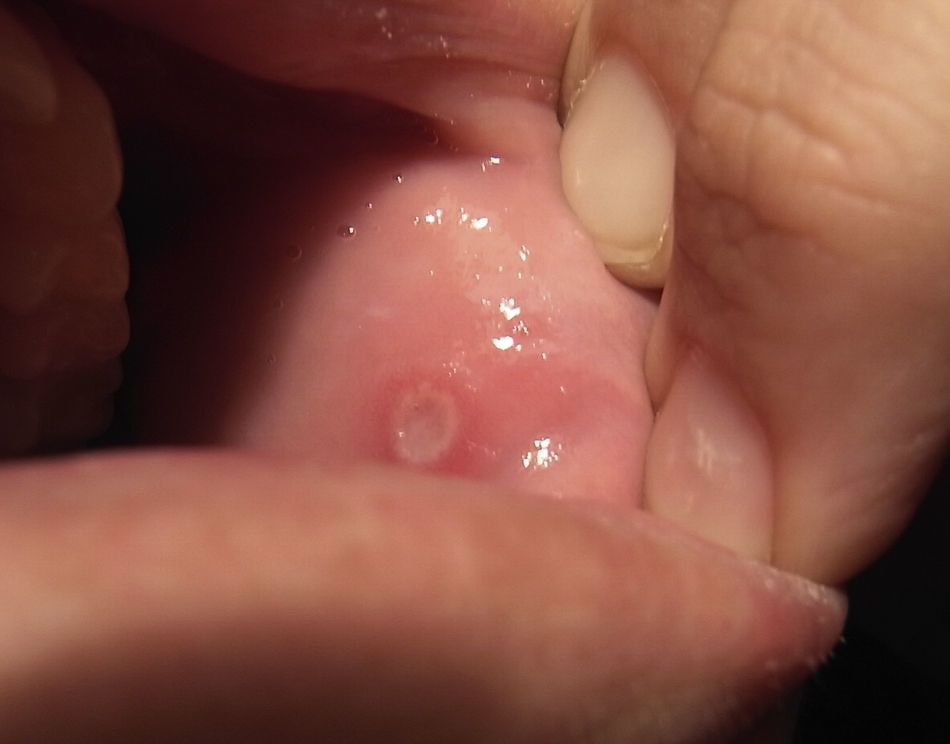
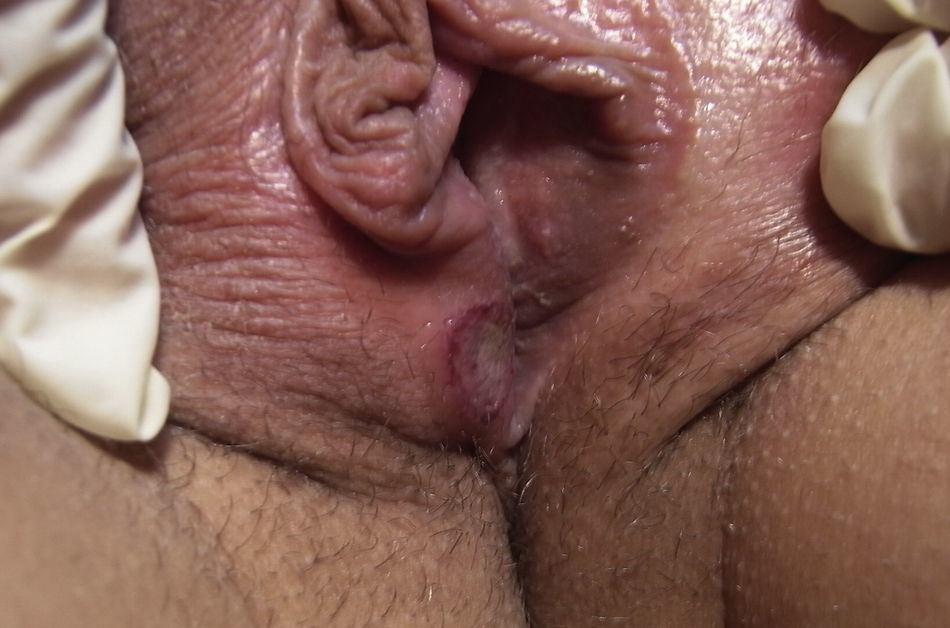
Un mes después del inicio de las lesiones cutáneas, y con estas aún sin resolver, la paciente acude de nuevo a la consulta por aftas orales (fig. 2) y úlceras genitales dolorosas (fig. 3), localizadas en introito y pared vaginal. La asociación de eritema nudoso, aftas orales y úlceras genitales despierta la sospecha de una enfermedad de Behçet por lo que se realiza la prueba de patergia mediante la inyección intradérmica de 0,1ml de suero fisiológico en el antebrazo. Hecha la lectura a las 24 h, el resultado es negativo al no aparecer una pústula donde se aplicó la inyección. Es preciso destacar que en los países occidentales esta prueba solo es positiva en el 30% de los casos.


La paciente es remitida al servicio de reumatología, que solicita una analítica que da el siguiente resultado: Hb 10,4; Hto 31,7; VCM 84,3; GR 3,76 millones/mm3; VSG 24; proteinograma, PCR, complemento C3, C4, enzima conversora de la angiotensina, TSH, T4 y bioquímica, normales; anticuerpos anti-DNA-nativo; ANCA y HLA-B27 negativos, e IgG HSV 1 y 2 positivos (25,6). Se pauta un tratamiento para el eritema nudoso con colchicina 0,5 mg/día y prednisona en dosis de 10 mg/día para la aftosis orogenital.
En los 12 meses transcurridos desde la aparición del eritema nudoso, la paciente ha tenido 3 brotes de aftas orales, un segundo brote de úlceras genitales y el eritema nudoso ha recurrido en 3 ocasiones.
El diagnóstico diferencial se realiza teniendo en cuenta cada manifestación. Las úlceras orales, con aftosis familiar recidivante, leucosis, agranulocitosis, eritema exudativo multiforme de Stevens-Johnson, colitis ulcerosa, enfermedad de Crohn, granulomatosis de Wegener, síndrome de Reiter, infección herpética, infección por el virus de inmunodeficiencia humana, lupus eritematoso sistémico, además de cáusticos, antisépticos, ácido acetilsalicílico, metotrexato y sales de oro.
Las úlceras genitales pueden deberse a infecciones herpéticas u otras infecciones de transmisión sexual. Sin embargo, la asociación de aftas orales y genitales recidivantes reduce el diagnóstico diferencial fundamentalmente a la infección herpética.
El eritema nudoso puede tener otras causas además de la enfermedad de Behçet, como la tuberculosis, la infección estreptocócica, fármacos, sarcoidosis, enfermedad de Crohn y vasculitis nodular.
El segundo caso (caso B) que presentamos es el de un varón de 31 años sin antecedentes personales de interés hasta el año 2005, en el que ingresa por una lesión seudotumoral inflamatoria en el tronco cerebral. Se resuelve sin secuelas con ampicilina y dosis elevadas de metilprednisolona.
El paciente se mantiene asintomático durante 5 años, hasta noviembre de 2010, cuando acude a la consulta por tos con expectoración hemoptoica, sudoración nocturna, discreto síndrome general y sin fiebre. La exploración es normal. Se solicita radiografía de tórax que es informada de aumento de densidad adyacente a la cisura menor del pulmón derecho, de difícil valoración radiológica, pero que sugiere como primera posibilidad una neumonía. Se inicia el tratamiento con amoxicilina-clavulánico 875 mg/125 mg, que hace remitir la tos y el síndrome general.
Tres semanas después acude a la consulta por tos seca, fiebre, sudoración nocturna, astenia, anorexia y adelgazamiento. En la radiografía de tórax de control persiste una imagen de opacidad similar al estudio previo.
Reinterrogado, el paciente refiere úlceras genitales y orales indoloras recidivantes de años de evolución y foliculitis de predominio en la región lumbar y las extremidades inferiores que nunca antes había consultado.
Se remite el paciente al hospital y en su ingreso presenta los datos siguientes:
- •
Exploración: PA 120/77, temperatura 39°C, FC 112, consciente, orientado, colaborador, aceptable estado general, normohidratado, eupneico, pulsos carotídeos simétricos, sin ingurgitación yugular, ni adenopatías. AC: rítmica, sin soplos; AP: buena ventilación en ambos campos pulmonares, sin roncus, ni sibilancias, ni crepitantes. Abdomen: blando depresible no doloroso, sin masas, ni megalias. MMII: pulsos presentes, sin edemas, ni datos de TVP. Neurológico: pupilas isocóricas normorreactivas, pares craneales normales, sin nistagmo, fuerza, marcha y sensibilidad normales; Romberg negativo, reflejo cutáneo plantar bilateral flexor, sin dismetrías. Mucocutáneo: aftas orales en el labio inferior, aftas genitales en bolsa escrotal derecha y foliculitis en miembros inferiores.
- •
Analítica: Hb 12, Hto 37, HCM 25, VCM 79, leucocitos 13.400 con 66% neutrófilos y 20% linfocitos; plaquetas 522.000; TP 1,28, TTPA 1,06, bioquímica, proteinograma, hormonas tiroideas normales, VSG 59, FR 22, proteína C reactiva 7,57, ANA y ANCA negativos. Hemocultivos negativos, auramina en esputo negativo. IF úlceras genitales negativo para herpes simple y varicela zóster.
- •
Radiografía de tórax: aumento de densidad adyacente a la cisura menor del pulmón derecho.
- •
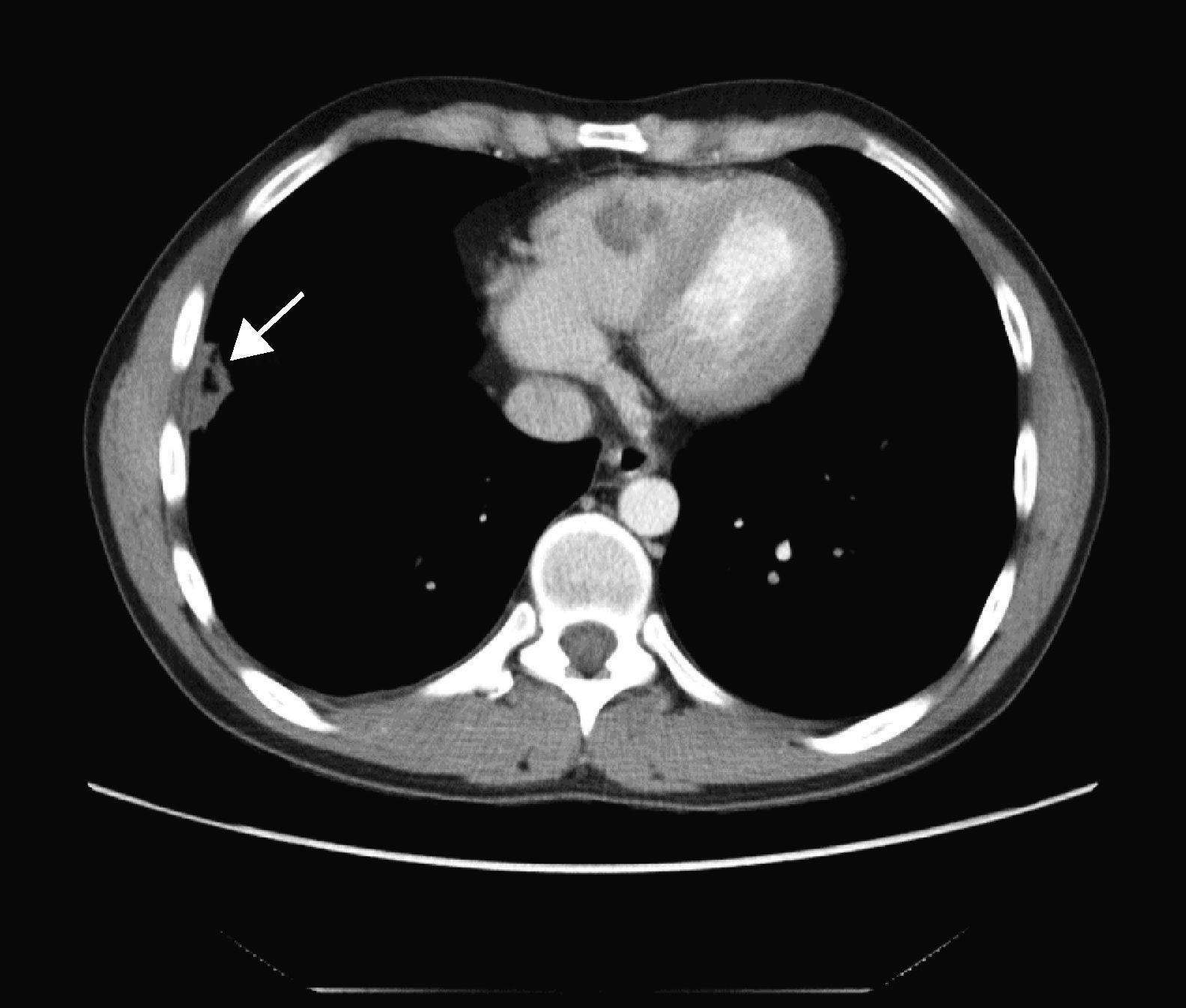
TAC toraco-abdominal: en el pulmón derecho se identifican 4 lesiones; la de mayor tamaño es muy organizativa y se encuentra adyacente a la cisura mayor-menor y otra lesión parenquimatosa en el lóbulo inferior adyacente a la cisura menor y que podría mostrar una pequeña cavitación (fig. 4). En el lóbulo inferior derecho aparecen 2 nódulos espiculados y una pequeña lesión parenquimatosa de localización muy posterior.
- •
Angio-TAC pulmonar: se visualizan trombos de aspecto crónico en la arteria del lóbulo inferior derecho y en arterias segmentarias basales de dicho lóbulo inferior y en alguna arteria segmentaria basal izquierda. También aparecen pequeñas lesiones parenquimatosas de localización periférica en base derecha, que pueden corresponder a infartos pulmonares, y una pequeña cantidad de fluido lobulado en la confluencia de las cisuras menor y mayor derechas.
- •
RMN cerebral: lesión en mesencéfalo en la región inferior de la porción compacta de la sustancia negra.
- •
Ecocardiograma: VI de tamaño y morfología normal, función sistólica normal de VI, flujo transmitral normal, VD de tamaño y morfología normales, función sistólica de VD normal, insuficiencia valvular tricuspídea de grado 1/4, ausencia de hipertensión pulmonar, sin derrame pericárdico.

El paciente ingresa por síndrome general, tos y fiebre. Se procede al aislamiento respiratorio por sospecha de tuberculosis. Tras el estudio analítico y radiológico se descarta la posibilidad infecciosa. Se realiza una angio-TAC de arterias pulmonares que muestra una tromboembolia pulmonar múltiple, por lo que se procede a la anticoagulación.
El paciente refiere haber tenido foliculitis cutánea, así como úlceras bucales de «toda la vida», y como úlceras genitales indoloras y recurrentes que nunca antes había consultado. En su ingreso hay presencia de estas lesiones, que se cultivan sin resultado. A la vista de estos datos se comenta el caso con Neurorradiología, que tras la revisión de imágenes del año 2005 y de las lesiones actuales de la RMN consideran el cuadro clínico muy indicativo de neuro-Behçet, y vasculitis cerebral secundaria a enfermedad de Behçet.
El diagnóstico diferencial en el caso de la tromboembolia pulmonar se establece principalmente con el lupus eritematoso sistémico, enfermedad de Buerger, síndromes antifosfolípidos y estados de hipercoagulabilidad. Por otra parte, el diagnóstico diferencial de la afección del sistema nervioso central en la EB es amplia y se haría con esclerosis múltiple, tumores cerebrales, compresión medular, hipertensión intracraneal idiopática benigna, parálisis aislada de los pares craneales, neurosífilis, neurosarcoidosis, meningitis crónicas y accidentes vasculares cerebrales.
DesarrolloLa enfermedad de Behçet fue descrita en 1937 por el médico turco Hulusi Behçet como un síndrome caracterizado por aftas orales, úlceras genitales y uveítis. Es más común en los países de la cuenca mediterránea oriental y Asia (antigua ruta de la seda) y muy frecuente en Turquía, con 80 a 370 casos por 100.000 habitantes. En países como Japón, Corea, China, Irán o Arabia Saudí varía entre 13,5-20 casos por 100.000 habitantes. En Norteamérica y norte de Europa es de 1 por 15.000 a 1 por 500.0001. En España la prevalencia es baja, estimada en 5-10 casos por 100.000 habitantes. En el área sanitaria de A Coruña la prevalencia es de 5,6 casos por 100.0002. La EB afecta a adultos jóvenes entre 20-40 años. Predomina en la mujer en las áreas menos prevalentes y en el hombre en las de mayor prevalencia3,4.
La EB es una enfermedad inflamatoria multisistémica crónica que evoluciona por brotes. Se ha incluido tradicionalmente en el grupo de las vasculitis de vasos pequeños por inmunocomplejos, junto a otras enfermedades como AR, LES, síndrome de Sjögren. Sin embargo, afecta a venas y arterias de cualquier tamaño, y en algunas lesiones como las cerebrales y las de seudoacné no hay daño directo de la pared vascular5.
La etiología de la EB es desconocida. Hay una predisposición genética que precisa de uno o varios desencadenantes desconocidos, pero que revisiones recientes apuntan hacia microorganismos (virus, micobacterias6, estreptocos7–10 y Helicobacter pylori11). Se ha asociado a genes HLA como el HLA-B51/B512 y el gen MICA13 (factor de riesgo genético para EB) y a otros genes no HLA como el gen 1-ICAM, gen sintetasa óxido nítrico endotelial, gen de TNf14–16, gen MEFV17 (este último presente también en la fiebre mediterránea familiar con la que se ha querido relacionar en el grupo de síndromes autoinflamatorios).
En relación con la patogenia, en el suero de los pacientes con EB se han detectado anticuerpos anticélulas endoteliales (AECA) tipo IgM anti-alfaenolasa. La alfaenolasa es una enzima intracelular que se expresa en la superficie de aquellas células hematopoyéticas estimuladas por polisacáridos. Una vez en la superficie celular se une a proteínas (proteínas de choque calórico, HSP) que interactúan con receptores específicos (toll-like receptores, TLR) para ser presentados al sistema inmunitario e inducir una respuesta mediada por los linfocitos T CD8; la secreción de citocinas proinflamatorias (interleucinas, factor de necrosis tumoral alfa, interferón gamma); la formación de autoanticuerpos y complejos inmunitarios locales dirigidos al endotelio, lo que provoca fenómenos de vasculitis y, de forma secundaria, trombosis. Los microorganismos anteriormente mencionados pueden expresar en su superficie alfaenolasa y otras proteínas y desencadenar mediante una reacción cruzada esta misma respuesta inmunitaria18.
Otros aspectos de la EB son la hiperreactividad neutrofílica directa o mediada por citocinas19,20; la participación predominante de los linfocitos helper Th 1 responsables de la hipersensibilidad retardada proinflamatorio21; y la disfunción endotelial, muy característica de la EB, en la que se ha implicado concentraciones altas de óxido nítrico y de homocisteína22–24.
Los pacientes con EB tienen un estado de hipercoagulabilidad resultado de un incremento de los factores de coagulación; factor VII, XI, Von Willebrand, ristocetina, antitrombina III, fibrinógeno y de un déficit de la APC (proteínaC reactiva), potente inhibidor de la coagulación25,26.
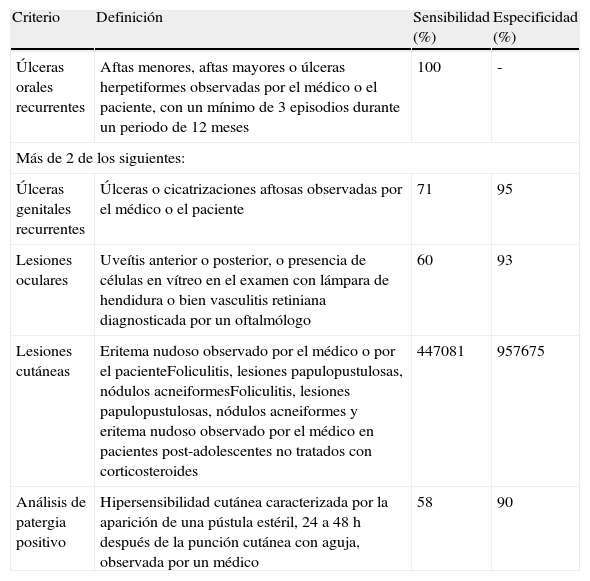
En cuanto al diagnóstico de la EB, no hay ninguna prueba de laboratorio patognomónica. El diagnóstico es clínico y se basa en los criterios del Grupo Internacional para el estudio de la EB del año 1990, según se expone en la tabla 1.
Criterios del Grupo Internacional para el estudio de la enfermedad de Behçet
| Criterio | Definición | Sensibilidad (%) | Especificidad (%) |
| Úlceras orales recurrentes | Aftas menores, aftas mayores o úlceras herpetiformes observadas por el médico o el paciente, con un mínimo de 3 episodios durante un periodo de 12 meses | 100 | - |
| Más de 2 de los siguientes: | |||
| Úlceras genitales recurrentes | Úlceras o cicatrizaciones aftosas observadas por el médico o el paciente | 71 | 95 |
| Lesiones oculares | Uveítis anterior o posterior, o presencia de células en vítreo en el examen con lámpara de hendidura o bien vasculitis retiniana diagnosticada por un oftalmólogo | 60 | 93 |
| Lesiones cutáneas | Eritema nudoso observado por el médico o por el pacienteFoliculitis, lesiones papulopustulosas, nódulos acneiformesFoliculitis, lesiones papulopustulosas, nódulos acneiformes y eritema nudoso observado por el médico en pacientes post-adolescentes no tratados con corticosteroides | 447081 | 957675 |
| Análisis de patergia positivo | Hipersensibilidad cutánea caracterizada por la aparición de una pústula estéril, 24 a 48 h después de la punción cutánea con aguja, observada por un médico | 58 | 90 |
Tomada de Graña Gil y Sánchez Meizoso40.
En el año 2006 se lleva a cabo una nueva revisión de los criterios internacionales para la EB (ICBD) y se le asignan 2 puntos a las aftas genitales recurrentes y a las lesiones oculares. Y un punto a las aftas orales recurrentes, lesiones cutáneas, lesiones vasculares (trombosis arteriales, venosas y aneurismas) y test de patergia positiva. Los pacientes con 3 o más puntos pueden ser diagnosticados de EB. La sensibilidad, la especificidad y la precisión de estos criterios (ICBD) es del 96,1, 88,7, y 93,8%, respectivamente27.
La positividad de la prueba de patergia en zonas no endémicas es muy baja. En España oscila entre un 13% en Galicia28, un 13,6% en Granada29 y un 60% en Canarias, esta última con una frecuencia similar a la de países de mayor prevalencia, como Turquía y Japón30.
Otras manifestaciones son las neurológicas, que afectan a menos del 30% de los pacientes y son más frecuentes en los hombres que en las mujeres. Suelen estar precedidas por otras manifestaciones de la enfermedad en 5-6 años. Las lesiones focales parenquimatosas (tracto piramidal, tronco cerebral, ganglios de la base, médula espinal), de peor pronóstico, y las complicaciones vasculares (ACV, aneurismas, hemorragia subaracnoidea, trombosis venosas) son las más comunes31,32.
La enfermedad arterial, en forma de estenosis, trombosis, aneurismas y hemorragias, aparece en un tercio de los pacientes y las arterias más afectadas son carótida, pulmonar, aorta, ilíacas, femorales y poplíteas. El IAM por arteritis coronaria es muy poco frecuente33,34.
La enfermedad venosa es más frecuente que la arterial y es aún más frecuente en pacientes con prueba de patergia positiva y/o uveítis. La trombosis venosa es una manifestación temprana de la EB. Las venas más afectadas son la cava superior, inferior, venas hepáticas (síndrome de Budd-Chiari), vena porta y seno dural. En un estudio con 2.319 pacientes turcos con EB, un 53,3% habían tenido una trombosis venosa superficial y un 29,8%, una trombosis venosa profunda35,36.
La artritis en la EB no es erosiva ni deformante; es asimétrica oligoarticular (2/3 casos), monoarticular (1/3 casos), y poliarticular en menos del 5%. La sacroilitis es rara37.
La afectación renal (proteinuria, hematuria, IR leve) y la cardiaca (carditis, arritmias ventriculares, trastornos de la conducción, prolapso mitral, fibrosis endocárdica) son poco frecuentes38,39.
Entre los trastornos gastrointestinales es preciso destacar las ulceraciones que afectan al íleon, ciego y colon ascendente, que pueden provocar perforación intestinal y, a nivel pulmonar, además de la afectación vascular, puede haber derrame pleural, estenosis bronquial, abscesos, bronquitis crónica y fibrosis pulmonar34.
El diagnóstico diferencial es amplio y varía en función de las manifestaciones clínicas del paciente. Los diagnósticos diferenciales de las aftas orales, genitales, el eritema nudoso, neuro-Behçet, y las manifestaciones vasculares ya han sido expuestos con cada caso clínico.
La presencia de úlceras perianales obliga a descartar la enfermedad de Crohn y la colitis ulcerosa. Las lesiones papulopustulosas simulan el acné. Otras causas de uveítis anterior son el síndrome de Reiter, la espondilitis anquilosante y la artritis crónica juvenil entre otras. La uveítis intermedia también aparece en la sarcoidosis y la vasculitis retiniana en la enfermedad de Eales.
La artritis (oligo-monoarticular) debe incluir las espondiloartropatías seronegativas (artritis reactivas, artropatía psoriásica, síndrome de Reiter, artritis asociada a la EII y la espondilitis anquilosante) y algunas colagenosis como el LES, además de la fiebre mediterránea familiar y el reumatismo palindrómico. Y, finalmente, las manifestaciones digestivas, con la enfermedad de Crohn y la colitis ulcerosa40.
Respecto al pronóstico, la EB es más grave en jóvenes, varones y en zonas endémicas. Las manifestaciones neurológicas, oculares, arteriales y venosas son las que tienen una mayor morbilidad y mortalidad4,41. Las manifestaciones oculares y neurológicas suelen progresar a pesar del tratamiento42. El retraso en el diagnóstico, frecuente en los países con baja prevalencia, incrementa la morbilidad y la mortalidad de los pacientes con EB43,44.
El tratamiento de la EB se basa en la administración de fármacos que atenúan los síntomas, en fármacos que modifican la enfermedad y en la incorporación novedosa de agentes biológicos antifactor de necrosis tumoral (TNF).
El tratamiento sintomático se realiza con corticoides tópicos para las lesiones orales, cutáneas, uveítis anterior; y con antiinflamatorios no esteroideos en caso de eritema nudoso y manifestaciones articulares.
Los fármacos que modifican el curso de la enfermedad son: colchicina a dosis 1-2 mg/día, indicada para tratar y prevenir las manifestaciones cutáneas y las articulares45; levamisol en dosis de 150 mg/día/3 días consecutivos por semana en el caso de lesiones mucocutáneas, articulares y uveítis leve46; corticoides orales para reducir la inflamación, en dosis que oscilan entre 5 mg/día para úlceras orales; y los 80 mg/día en el caso de uveítis posterior o meningitis aséptica. Habitualmente se emplean en asociación con fármacos citotóxicos. En este grupo se sitúan la azatioprina, ciclosporina, ciclofosfamida, metotrexato e interferón alfa. Todos son eficientes en las formas graves de la enfermedad, especialmente en las lesiones oculares, neuro-Behçet, manifestaciones gastrointestinales graves, vasculitis pulmonar y complicaciones vasculares graves47.
La ciclosporina A es efectiva en dosis de 5 mg/kg por vía oral; asociada a citotóxicos ha demostrado su eficacia en manifestaciones oculares. La vasculitis retiniana responde mejor a la combinación de pulsos de ciclofosfamida con azatioprina que otras pautas48,49.
Los agentes biológicos anti-TNF (factor de necrosis tumoral); interferón alfa, infliximab, etanercept, adalimumab, rituximab, son la última incorporación al tratamiento de la EB. Se utilizan cuando los fármacos citotóxicos son ineficientes. Sin embargo, carecen de grandes series y no tienen estudios aleatorios controlados excepto para etanercept en las manifestaciones mucocutáneas y rituximab en las oculares. Su eficacia a largo plazo es desconocida, excepto para el interferón alfa en las manifestaciones oculares, que ha demostrado una buena respuesta en un 92% de los casos50.
Mención aparte es el papel de la anticoagulación en la trombosis venosa profunda como tratamiento clásico que ha de mantenerse durante años. En las trombosis de grandes vasos la tendencia actual creciente es asociar a la anticoagulación, citotóxicos (ciclofosfamida) y prednisolona, que aumentan la eficacia de la anticoagulación y permitirían plantear su suspensión después de un par de años47.
ConclusionesLa EB es una enfermedad multisistémica crónica con afectación inflamatoria vascular en algunas lesiones, pero no en otras, como las lesiones parenquimatosas cerebrales y las de seudoacné, por lo que su inclusión en el grupo de las vasculitis está actualmente en discusión. La causa de la EB es desconocida. Hay una predisposición genética que se ha demostrado por su asociación a genes HLA (HLA B51/B5, gen MICA) y otros no HLA (gen 1-ICAM, gen TNF, gen MEFV); y además precisa de otros factores que las últimas investigaciones orientan hacia la participación de microorganismos como estreptococos, micobacterias, virus, y Helicobacter pylori.
Los criterios diagnósticos de la EB fueron revisados en el año 2006 por el Grupo Internacional para la revisión de los criterios internacionales de la EB (ICBD). La novedad de dicha revisión es la inclusión de las lesiones vasculares (trombosis arteriales, venosas, aneurismas) como criterio diagnóstico, además de los criterios ya conocidos como aftosis orogenital, lesiones cutáneas, lesiones oculares y test de patergia positivo. Se adjudica a cada manifestación clínica una puntuación, de forma que con 3 o más puntos se diagnostica EB. La incorporación de las lesiones vasculares como criterio diagnóstico es importante en países como España, que tienen una prevalencia muy baja del test de patergia positivo (13%). Es preciso señalar el empleo de fármacos anti-TNF (infliximab, etanercept, adalimumab, rituximab) en el caso de contraindicación, intolerancia o fracaso de tratamientos previos con corticoides e inmunosupresores.
Y, por último, es importante estar familiarizados en atención primaria con las características de una enfermedad que esconde su diagnóstico en una variedad de síntomas que hay que asociar para descubrirla, además de excluir otros procesos con manifestaciones similares, ya que cualquier retraso aumenta la morbilidad y mortalidad de los pacientes con la EB.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
