




La hemoglobina (Hb) es la proteína responsable del transporte de oxígeno a los tejidos del organismo y está formada por cuatro cadenas de globina y cuatro grupos hemo. Una persona adulta sana, posee 2 genes beta y 4 genes alfa, que codifican la síntesis de las 3 fracciones de Hb1,2:
1) Hb A, formada por 2 cadenas α y 2 β. Representa el 96-98% del total de Hb.
2) Hb A2, formada por 2 cadenas α y 2 δ. Representa el 2,5-3,5% del total de Hb.
3) Hb F, formada por 2 cadenas α y 2 cadenas βδ. Supone menos del 1,5% del total de Hb.
Las talasemias son un grupo heterogéneo de trastornos hereditarios cuya característica común es un defecto en la síntesis de una o varias de las cadenas de globina3. Cada talasemia recibe el nombre de la cadena que deja de sintetizarse. Las más comunes son: a) la β-talasemia: resultado de la falta de síntesis de las cadenas β, con un exceso de cadena α; b) la α-talasemia: resultado de la falta de síntesis de las cadenas α, con un exceso de cadenas β y c) la δ β-talasemia, cuando falta más de una cadena (β δ).
La disminución de la síntesis de las cadenas de globina provoca un desequilibrio entre cadenas alfa y beta, provocando una acumulación anormal de la globina excedente. Estos precipitados intracelulares son los responsables de la destrucción precoz de los eritroblastos, tanto en la propia médula ósea (eritropoyesis ineficaz), como en la sangre periférica (hemólisis). Los hematíes presentan un volumen corpuscular medio (VCM) y una hemoglobina corpuscular media (HCM) disminuidos.
Se han descrito cientos de mutaciones y deleciones en los genes de la globina, que pueden dar origen a diferentes formas de talasemias2.
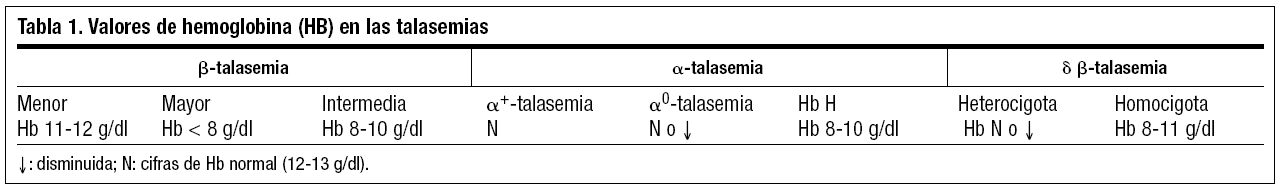
El grado de severidad de la anemia en las diferentes formas de talasemia depende del tipo de mutación y del carácter homocigoto o heterocigoto (tabla 1). Desde el punto de vista clínico, las talasemias pueden ser asintomáticas (talasemia silente), pueden ser clínicamente manifiestas, (anemia hemolítica) y llegar incluso a ser incompatible con la vida (hidropesía fetal).
BETA-TALASEMIA
Las β-talasemias son el resultado de la falta de síntesis de las cadenas β de globina, con un exceso de cadenas α-globina. El exceso de cadena α hace que se formen tetrámeros insolubles, que precipitan y dañan la membrana de los glóbulos rojos, provocando eritropoyesis ineficaz y hemólisis. La mayoría de casos de β-talasemia se deben a mutaciones genéticas1 (cambios en las bases nitrogenadas de la cadena b) que afectan al funcionamiento del ARN. Podemos distinguir diferentes formas:
β-talasemia menor (o rasgo talasémico)
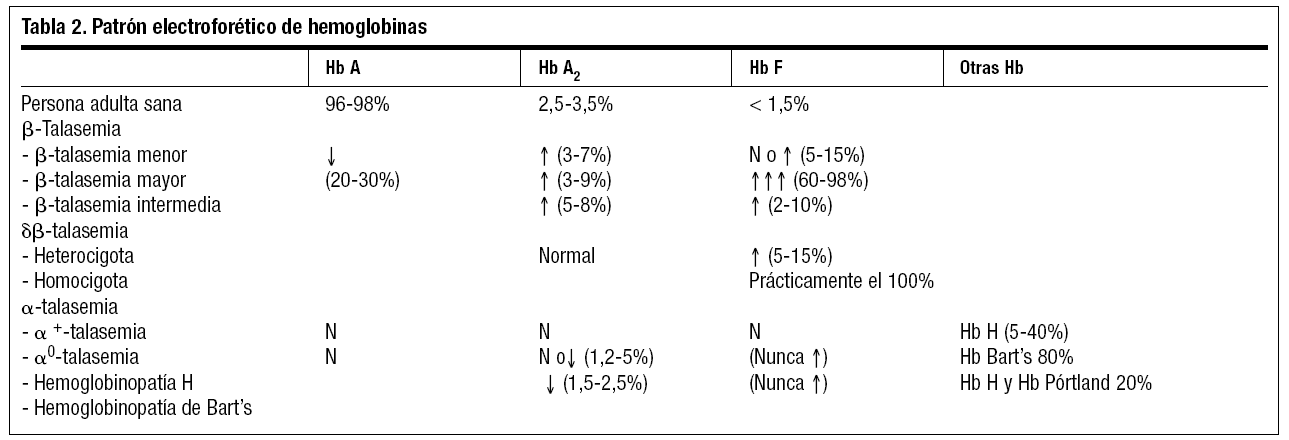
Es el resultado del estado heterocigoto para una mutación del gen β. Es la forma más frecuente de β-talasemia en España, así como en los países de la ribera mediterránea1,2,4. Las personas portadoras del rasgo talasémico generalmente no presentan manifestaciones clínicas, y su hallazgo suele ser casual con motivo de algún examen hematológico de rutina. Su expresividad más característica es la pseudopoliglobulia microcítica (aumento de la cifra de hematíes con VCM < 80 FL) sin anemia o con anemia muy discreta (Hb 10-12 g/dl), con una amplitud de distribución eritrocitaria (ADE) normal o levemente elevada, y los reticulocitos pueden estar elevados. En el estudio de electroforesis de Hb, encontramos un aumento de Hb A2, con Hb F normal, (aunque puede aumentar hasta 5%).
β-talasemia mayor
Resultado del estado homocigoto para la mutación de los dos genes de la cadena β. Dependiendo de la mutación, la cantidad de cadena β que se sintetiza es nula o muy escasa. Su expresividad clínica es variable, pero generalmente intensa. Su forma más severa es la anemia de Cooley, una anemia hemolítica congénita muy grave. La enfermedad comienza a manifestarse a parir del quinto-sexto mes de vida. Durante los 2 primeros años de vida, estos pacientes presentan palidez, irritabilidad, alteraciones del sueño, rechazo del alimento e infecciones de repetición. La anemia es muy intensa y progresivamente aparecen complicaciones como son esplenomegalia y hepatomegalia. Presentan también alteraciones en el sistema óseo con deformidades (huesos largos, delgados) y fracturas espontáneas. Son típicas las alteraciones radiológicas como el "cráneo en cepillo".
En el hemograma encontramos una anemia microcítica intensa (Hb < 8 g/dl), eritroblastos en sangre periférica, y los reticulocitos están ligeramente elevados. En el estudio electroforético, el 60-98% de la Hb que aparece es HbF, hay una pequeña cantidad de Hb A2 y un porcentaje variable de Hb A.
El requerimiento transfusional es constante y las complicaciones de la sobrecarga férrica5 secundaria a las transfusiones son la causa de muerte en estos pacientes, generalmente antes de los 25 años4.
β-talasemia intermedia
Corresponde a formas de expresividad clínica de diferente intensidad, aunque siempre presenta anemia (Hb ente 8-10 g/l) con síndrome hemolítico y esplenomegalia. En el estudio electroforético aparece un ligero aumento de Hb A2 (5-8%), y Hb F > 2%. Estos pacientes no suelen depender de transfusiones.
DELTABETA-TALASEMIA
Se caracteriza por un defecto en la síntesis de cadenas β y δ. Es relativamente frecuente en las regiones mediterráneas. La expresividad clínica depende del carácter heterocigoto u homocigoto de la mutación:
δβ-talasemia heterocigota
Se caracteriza por ausencia de manifestaciones clínicas y en el hemograma hallamos: Hb normal o algo disminuida, aumento del número de hematíes, microcitosis y ADE aumentada. La Hb A2 es normal, con aumento de Hb F (5-15%).
δβ-talasemia homocigota
Presenta las manifestaciones clínicas de una talasemia intermedia. Presenta una anemia de 8-11 g/dl, con intensa microcitosis y prácticamente el 100% es Hb F. No requiere transfusiones.
ALFA-TALASEMIA
Las deleciones de genes de globina constituyen el mecanismo más frecuente de la alfa-talasemia2. Hay diferentes formas:
α+-talasemia (portador silente)
Aparece cuando se produce la deleción de un solo gen. Son personas sanas, sin manifestaciones clínicas. El patrón de electroforesis de Hb es normal. Únicamente puede detectarse mediante estudio del ADN. No precisa tratamiento.
α0-talasemia o α-talasemia menor (rasgo talasémico)
Aparece si la deleción es de dos genes. Generalmente no presenta manifestaciones clínicas, y su expresividad clínica es superponible a β-talasemia menor. El patrón electroforético es normal, con Hb A2 normal o discretamente disminuida (1,5-2,5%), pero nunca elevación de Hb F. No precisa tratamiento.
Hemoglobinopatía H o talasemia de grado intermedio
Aparece si la deleción es de tres genes. Su expresividad clínica es superponible a la β-talasemia intermedia. Es habitual que esta anemia hemolítica aumente si se asocia con determinadas infecciones (gripe, anginas, infecciones urinarias) o con determinados fármacos3, (antipalúdicos, sulfamidas, nitrofurantoína, vitamina K, ácido nalidíxico etc.), capaces de inducir un estrés oxidante en el eritrocito y provocar episodios de hemólisis. Por este motivo, requiere controles médicos periódicos. En la electroforesis encontramos Hb H (5-40%), una cantidad variable de Hb Bart's, la Hb A2 está disminuida (1,5-2,5%), pero nunca aumenta la Hb F. En algunas ocasiones precisa de transfusiones.
Hemoglobinopatía de Bart's o a-talasemia mayor
Aparece si se produce la deleción de los cuatro genes. Casi exclusivo de países del sudeste asiático (China, Filipinas, Tailandia). Es una forma muy grave, incompatible con la vida. La muerte se produce antes o inmediatamente después de nacer. La electroforesis de Hb muestra un 80% de Hb de Bart's, 20% de Hb H y Hb Pórtland.
DISTRIBUCIÓN Y FRECUENCIA
La frecuencia de la talasemia es variable en las diferentes zonas geográficas2, y las diferentes modalidades de la patología presentan también una distribución geográfica heterogénea:
1) En poblaciones del área Mediterránea, las frecuencias varían ente el 1 y 30%, predominando la β-talasemia. La mayor incidencia de β-talasemia en determinadas áreas está marcada por la selección positiva ejercida por el paludismo endémico2,6, (la hemoglobinopatía ejerce un efecto protector frente al parásito).
2) En Oriente Medio, sudeste Asiático y China, las frecuencias varían entre el 5 y el 40%, con predominio de alfa-talasemia.
En España6, la prevalencia de b-talasemia es más baja que en el resto de los países mediterráneos, y con una distribución irregular4. Mientras que entre la población del País Vasco destaca la ausencia de talasemia, hay zonas, como la isla de Menorca7, donde la prevalencia de β-talasemia menor se sitúa entre el 2,6-7%. Los estudios efectuados en distintas áreas geográficas arrojan una prevalencia promedio de β-talasemia de 0,4%, lo que supone un paciente con β-talasemia por cada 250 habitantes6. Para determinar la prevalencia de α-talasemia, se han hecho estudios aplicando métodos moleculares en el ADN de sangre de cordón umbilical de recién nacidos6, y se observa una prevalencia de alelos alfa-talasémicos entre el 1,25-2,2%, lo que supone entre 3 y 6 pacientes por cada 250 habitantes.
DIAGNÓSTICO
Los portadores pueden identificarse, la mayoría de las veces, mediante un examen hematológico elemental, incluyendo Hb, sideremia y frotis de sangre periférica, así como la determinación de Hb A2 y Hb F. El estudio de electroforesis de Hb y la cuantificación de las hemoglobinas sirve para confirmar la existencia de talasemia (tabla 2), aunque una cuantificación normal de Hb A2 y Hb F no excluye el diagnóstico, (en α-talasemia pueden ser normales)
La confirmación última del diagnóstico y el tipo específico de la talasemia se hace por estudio de cadenas de globina y ADN molecular. El análisis del ADN tiene un valor primordial en el diagnóstico prenatal. Mediante la reacción en cadena de la polimerasa se puede amplificar el ADN obtenido por biopsia de las vellosidades placentarias o por amniocentesis.
TRATAMIENTO
Las talasemias que son asintomáticas o las que cursan con escasas manifestaciones clínicas no necesitan tratamiento. Sin embargo, las formas clínicamente manifiestas sí precisan de tratamiento y las posibilidades terapéuticas son las siguientes:
1) Transfusión de sangre con la periodicidad necesaria para mantener las cifras de Hb por encima de 10 g/l. Es el tratamiento básico, y debe acompañarse de la administración subcutánea de deferroxamina para evitar la sobrecarga férrica5 (hemocromatosis secundaria).
2) Suplementos de ácido fólico.
3) La esplenectomía debe aconsejarse en aquellos pacientes con hiperesplenismo intenso o compresión de órganos vecinos por esplenomegalia.
4) El trasplante de médula ósea4 es hoy en día el único tratamiento que puede resultar curativo, con una tasa de supervivencia media superior a 5 años del 75-90% y una mortalidad del 10-20%.
En un futuro próximo, es posible que mediante técnicas de ingeniería genética se consiga implantar genes normales en los precursores de los eritroblastos, corrigiendo el déficit genético.
También el diagnóstico prenatal8 podría ayudar en el tratamiento de la beta-talasemia. La evaluación prenatal del antígeno de histocompatibilidad HLA, puede:
1) Identificar a los fetos que desarrollarán β-talasemia y que pueden ser tratados con trasplante de médula ósea de un miembro de la familia.
2) Identificar a los fetos con histocompatibilidad para el tratamiento de la β-talasemia de sus hermanos, mediante cultivo de células de cordón umbilical y un futuro trasplante.
¿CON QUÉ PROBABILIDAD SE TRANSMITE ESTE TRASTORNO DE PADRES A HIJOS?
La talasemia es un trastorno genético que se transmite con un patrón de herencia autosómica dominante. Dado que la forma más prevalente en España es la β-talasemia, voy a presentar un ejemplo del consejo genético que podemos dar a nuestros pacientes afectados por dicho trastorno hereditario:
1) Si uno de los miembros de la pareja presenta β-talasemia menor, cada uno de los hijos tendrá una probabilidad del 50% de heredarlo y el 50% de nacer totalmente sanos. Ninguno de los hijos tendrá talasemia mayor.
2) Si ambos progenitores presentan β-talasemia menor, en cada embarazo la probabilidad de que el hijo tenga la sangre normal es del 25%, la probabilidad de que sea portador de β-talasemia menor es del 50% y un 25% de probabilidades de tener una β-talasemia mayor.
3) Si ninguno de los progenitores es portador de β-talasemia, no se puede transmitir a sus descendientes.
COMENTARIO
La β-talasemia es la forma más frecuente en España, siendo la prevalencia menor que en el resto de los países mediterráneos. Pero debido a los movimientos migratorios que se están produciendo en los últimos años en nuestro país, es de esperar que el mapa de distribución geográfica de la talasemia, sus tasas de prevalencia y su espectro mutacional sufran variaciones.
Ante un hallazgo de microcitosis con o sin anemia, hay que hacer un adecuado diagnóstico diferencial. Las causas más frecuentes de microcitosis (VCM < 83 fl) son: ferropenia, procesos inflamatorios crónicos, talasemia y anemia sideroblástica. Confundir microcitosis y ferropenia es un error que vemos con muchísima frecuencia en la práctica médica diaria y a todos los niveles asistenciales. Las microcitosis no deben ser tratadas con hierro mientras no se compruebe un déficit del mismo (disminución de sideremia, con índice de saturación de la transferrina inferior al 16% y disminución de ferritina sérica), ya que esto supondría someter al paciente a un tratamiento prolongado e inútil, y a una nociva sobrecarga de hierro. El médico debe sospechar una talasemia por los hallazgos analíticos: anemia, microcitosis, aumento de la cifra total de hematíes, ADE normal o ligeramente elevada, y los parámetros relacionados con el metabolismo del hierro (sideremia, índice de saturación de transferrina y ferritina) normales.
Los antecedentes familiares de microcitosis o de anemia microcítica son datos que el médico de familia debe tener en cuenta, ya que le pueden servir de ayuda en el diagnóstico diferencial y orientar al diagnóstico de talasemia.
Si bien muchas formas de talasemias son asintomáticas y no precisan de tratamiento, su diagnóstico es importante. El médico de Atención Primaria debe explicar al paciente las características de la enfermedad y sobre todo dar consejo genético para evitar la aparición de formas graves. La β-talasemia mayor es una enfermedad con gran repercusión en la calidad de vida, y sólo curable con trasplante de médula ósea; parece lógico aprovechar la relativa facilidad con que se pueden identificar a los portadores heterocigotos, y darles un adecuado consejo genético para prevenir la aparición de homocigotos. Cuando se detecta un paciente con talasemia, es importante hacer el estudio de los familiares (hijos, hermanos o los padres si están en edad de reproducción) y sus posibles parejas para detectar casos desconocidos. El consejo genético es fundamental en aquellas zonas de elevada prevalencia de talasemia y en zonas donde el grado de endogamia sea alto. La campaña de prevención de talasemia mayor llevada a cabo en Menorca7 fue muy eficaz, y tras 10 años de campaña, no nació ni un sólo niño con talasemia mayor.
Ante la sospecha de una talasemia, el médico de familia solicitará al laboratorio la determinación de las Hb A2 y Hb F, si bien es cierto que existen muchas diferencias entre los Servicios de Salud de las diferentes Comunidades Autónomas y, en algunas de ellas, esta determinación no puede ser solicitada desde Atención Primaria.
Serán motivo de derivación al servicio de hematología todos aquellos casos que:
1) Cursen con anemia severa
2) Precisen confirmación diagnóstica, porque el médico sospecha una posible talasemia pero el laboratorio de referencia no pueda determinar la Hb A2 y Hb F solicitada desde Atención Primaria.
3) Los familiares y las parejas de los portadores, si el médico de Atención Primaria no puede hacer dicho estudio por falta de accesibilidad a las pruebas de laboratorio.
4) Si se sospecha de una posible α-talasemia,
Como resumen, quiero destacar:
1) El médico de Atención Primaria debe mantener un alto índice de sospecha, (ante determinados hallazgos de laboratorio) y diagnosticar las posibles talasemias.
2) Reafirmar la labor del médico de familia como primer responsable de la prevención, mediante el consejo genético de los portadores heterocigotos.
Correspondencia: M.C. Goñi Murillo.
C/ San Miguel 12.
31160 Orkoien. Navarra.
Correo electrónico: cgonimur@cfnavarra.es
Recibido el 11-09-2006; aceptado para su publicación el 19-10-2007.
