






INTRODUCCIÓN: IMPORTANCIA DE LOS ESTUDIOS EXPERIMENTALES EN LA INVESTIGACIÓN CLÍNICA
El objetivo de toda labor de estudio e investigación, en general, es ampliar los conocimientos existentes sobre un tema y llegar a conclusiones válidas que aporten alternativas, y en el mejor de los casos, mejoras, sobre los conocimientos existentes a priori. La investigación clínica, en particular, pretende resolver cuestiones planteadas en cada una de las etapas del proceso asistencial, con el objetivo de mejorar la práctica clínica diaria y la calidad de la atención prestada a los pacientes. A partir de este objetivo, los diversos estudios epidemiológicos realizados por los investigadores aportan diferentes niveles de conocimiento ante cada una de las cuestiones planteadas.
Los estudios analíticos, tal como se indicó en el capítulo anterior, estudian la relación existente entre diferentes variables, permitiendo establecer relaciones causales1. Dentro de éstos, los estudios experimentales, al ser el investigador el que controla el factor de estudio y el resto de condiciones que rodean a la investigación, se consigue disminuir la posibilidad de que otros factores influyan sobre los resultados, y, por tanto, proporcionan una mayor confianza sobre la veracidad de las conclusiones obtenidas. En otras palabras, los estudios experimentales, y de forma más específica los ensayos clínicos aleatorios (ECA), aportan un nivel más alto de evidencia2 que el resto de diseños epidemiológicos descritos, encabezando las listas de cualquier clasificación de la calidad de la evidencia. La importancia de este diseño en el campo de la biomedicina se aprecia, por ejemplo, al observar que la mayoría de las recomendaciones terapéuticas y guías de práctica clínica se basan en las pruebas proporcionadas por ECA, o que las autoridades sanitarias obligan a la realización de un ECA para demostrar la eficacia y seguridad de un nuevo fármaco antes de su comercialización. Constituyen el tipo de diseño que nos acerca, de forma más directa, a la noción de causalidad entre una intervención y unos resultados concretos.
El diseño experimental más importante es el ECA, sobre el que hablaremos durante todo este capítulo, pero también existen otros tipos de estudios experimentales. La prueba o ensayo de campo se realiza sobre sujetos sanos, que aún no han adquirido la enfermedad, o sobre sujetos que están en riesgo de adquirirla, y estudia factores preventivos, como es el caso de la administración de vacunas, o el seguimiento de dietas. Generalmente se utilizan para el estudio de enfermedades muy frecuentes, o muy graves. La intervención se realiza de forma individual, y por tanto los sujetos son aleatorizados de forma individual a los diferentes grupos de estudio. La información se recoge sobre la comunidad misma, en el ambiente epidemiológico natural de los sujetos, sin necesidad de una institución cerrada específica para realizar el estudio.
Los ensayos comunitarios o de intervención3 son estudios experimentales que se emplean para probar hipótesis. Se realizan, al igual que el ECA, sobre un grupo experimental y uno de control, aunque la diferencia con éste radica en que los grupos de intervención y control no son individuos elegidos al azar, sino comunidades completas. De este modo, la asignación a los grupos no es aleatoria, sino que se eligen comunidades enteras por razones de conveniencia del investigador, o de las autoridades interesadas en el estudio. Son diseños buenos para probar hipótesis de intervenciones educativas, para evaluar sistemas de gestión y organización de la atención médica, o para abordar enfermedades crónicas no transmisibles asociadas a condiciones socioculturales o a estilos de vida.
EL ENSAYO CLÍNICO ALEATORIO: FASES
Un ECA es un experimento planificado en el que, de forma prospectiva, se comparan dos o más intervenciones preventivas, curativas o rehabilitadoras, que son asignadas de forma individualizada y aleatoria a un grupo de pacientes, con el objetivo de estudiar la eficacia y/o seguridad de dichas intervenciones en el hombre4. Tanto la selección de los sujetos como los períodos de tratamiento y seguimiento han de tener lugar simultáneamente en todos los grupos (grupos de intervención y grupos control).
En la tabla 1 se resumen las condiciones básicas de un ECA.

Cuando la intervención estudiada es un nuevo fármaco que se pretende introducir en el mercado, ha de superar un complejo proceso que requiere gran cantidad de recursos y tiempo hasta obtener la aprobación de las agencias reguladoras de cada país3. En primer lugar, los compuestos pasan por una fase de investigación preclínica, durante la cual se realizan estudios de laboratorio y en animales para detectar actividad biológica ante una determinada enfermedad, y realizar una evaluación previa de su seguridad. Si el fármaco logra superar satisfactoriamente esta fase, y cuenta con el permiso de las autoridades sanitarias para estudiar el producto en humanos, pasaría a la etapa de desarrollo clínico. Dentro de esta etapa el fármaco pasará por cuatro fases. Durante la fase I se realizan estudios no controlados en los que el producto se administrará a voluntarios sanos para estudiar el perfil de seguridad del fármaco, los rangos de seguridad de la dosificación y algunos datos farmacológicos relativos a la absorción, distribución, metabolización, excreción y duración de su acción. En esta fase también se realiza la denominada prueba de concepto, para determinar si existen indicios razonables de que el fármaco puede ser eficaz para la indicación seleccionada. Si el fármaco se considera razonablemente seguro para el hombre pasará a la fase II, donde se realizarán estudios no controlados y ECA controlados con placebo. El fármaco de administrará a un número mayor de voluntarios (pacientes potenciales o voluntarios sanos) con la finalidad de confirmar la seguridad, evaluar la eficacia en humanos a corto plazo y determinar algunos parámetros farmacocinéticos y farmacodinámicos como la dosificación, para ensayos posteriores. Una vez probada la eficacia a corto plazo el fármaco pasa a la fase III, donde se determinará la eficacia y seguridad a medio plazo, evaluando la relación beneficio/riesgo en comparación con otras alternativas terapéuticas disponibles, mediante la realización de ECA sobre muestras de 1.000 a 3.000 pacientes. Si el fármaco logra superar todas estas fases se lanza al mercado, y a partir de aquí entraría en una fase IV, donde se realizan ECA y estudios observacionales para obtener un mayor conocimiento sobre su perfil de seguridad, sobre nuevas indicaciones o vías de administración, y sobre su eficacia real en las condiciones habituales de uso.
METODOLOGÍA DEL ENSAYO CLÍNICO
Selección de los participantes y tamaño de la muestra
El sujeto o participante de un ECA es la persona sana o enferma que participa en el mismo después de haber otorgado libremente su consentimiento informado. Los motivos por los que un sujeto decide participar libremente en un ECA pueden ser muy variados: por la oportunidad de contribuir al avance de la ciencia y el beneficio de la colectividad, por tener un acceso preliminar a medicamentos nuevos, para obtener acceso a médicos y especialistas de prestigio, o para recibir la información y la estrecha atención médica que no cubre su plan de seguro.
A la hora de seleccionar a los participantes hay que tener en cuenta, por un lado, cuál es la población diana sobre la que se desean extrapolar los resultados de nuestro estudio, y por otro, cómo seleccionar la población experimental sobre la que se realizará el estudio, una población definida por una serie de criterios especificados a priori. Es muy importante el establecimiento de unos adecuados criterios de inclusión, que definirán el perfil de los sujetos susceptibles de participar en el estudio y los más adecuados para el objetivo de éste (sexo, edad, lugar de residencia, patologías actuales o pasadas, medicación actual, etc.). Estos criterios permiten definir una población homogénea sobre la que la intervención estudiada estaría igualmente indicada y potencialmente podría suponer el mismo nivel de riesgo o de beneficio. Además, es preciso establecer unos criterios de exclusión, que impidan participar en el estudio a sujetos en los que una de las alternativas de intervención pueda ser preferible a la otra, o aquellos en los que cualquiera de las intervenciones pueda estar contraindicada, pueda presentar interacciones, o presenten alguna característica que pueda comprometer la participación en el estudio o afectar a la variable de interés.
Es conveniente, a la hora de definir los criterios de selección de los participantes4,5, alcanzar un buen equilibrio entre unos criterios de selección estrictos, que conducirían a una muestra muy homogénea, aumentando la validez interna del estudio, o unos criterios más laxos, más amplios, que permiten seleccionar una muestra más representativa de la población diana y, por tanto, una mayor posibilidad de generalizar los resultados obtenidos (mayor validez externa). Criterios muy estrictos hacen que la muestra de nuestro estudio se aleje de la población general y que limite la capacidad de extrapolar los resultados al resto de sujetos que son potenciales pacientes susceptibles de recibir la intervención en el futuro. Sin embargo, criterios muy amplios, al conducir a muestras más heterogéneas hacen que sea más difícil detectar respuestas significativas al tratamiento, y requieren experimentar sobre muestras de sujetos más amplias.
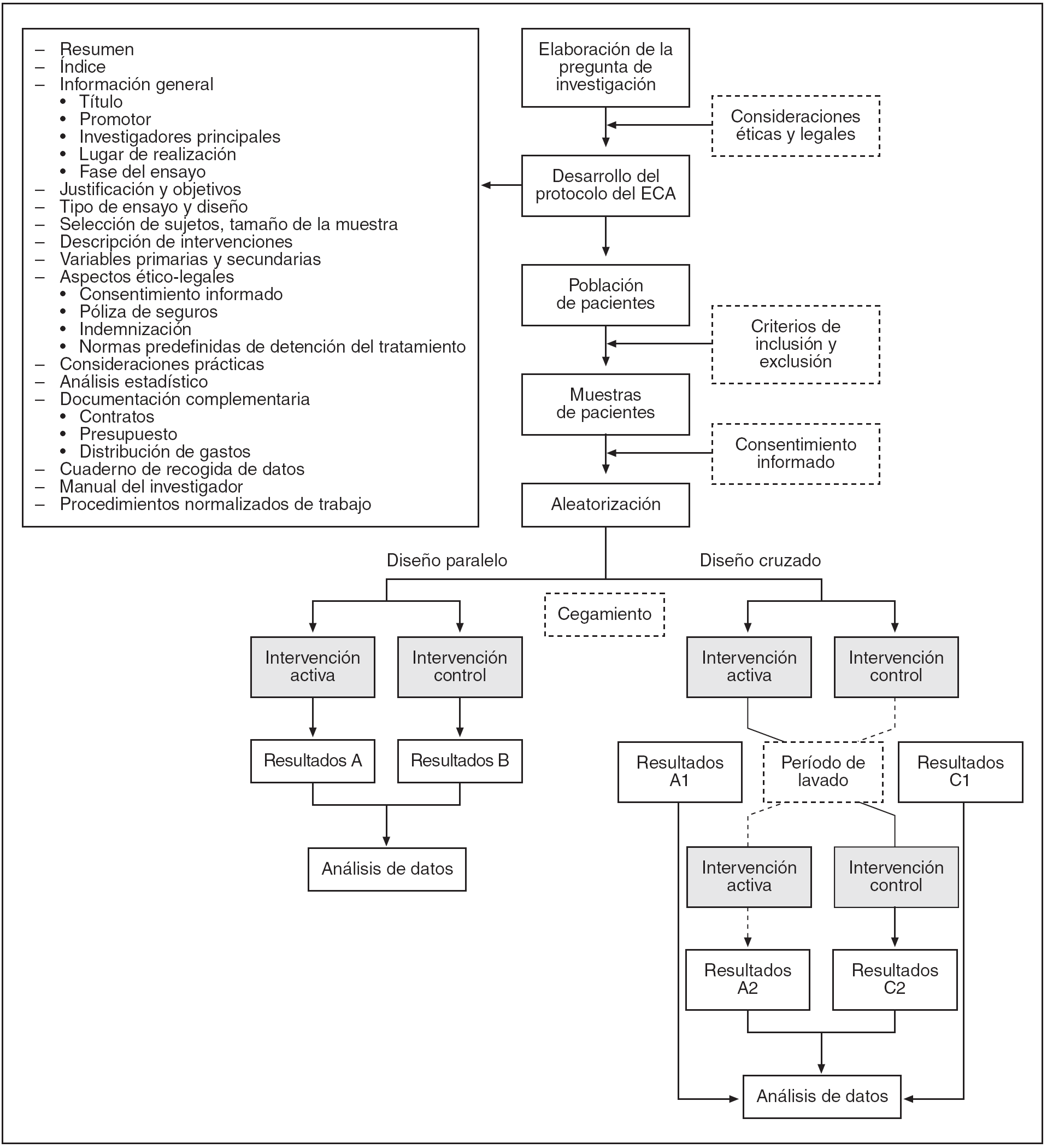
En la figura 1 se muestran las etapas de la elaboración de un ECA.
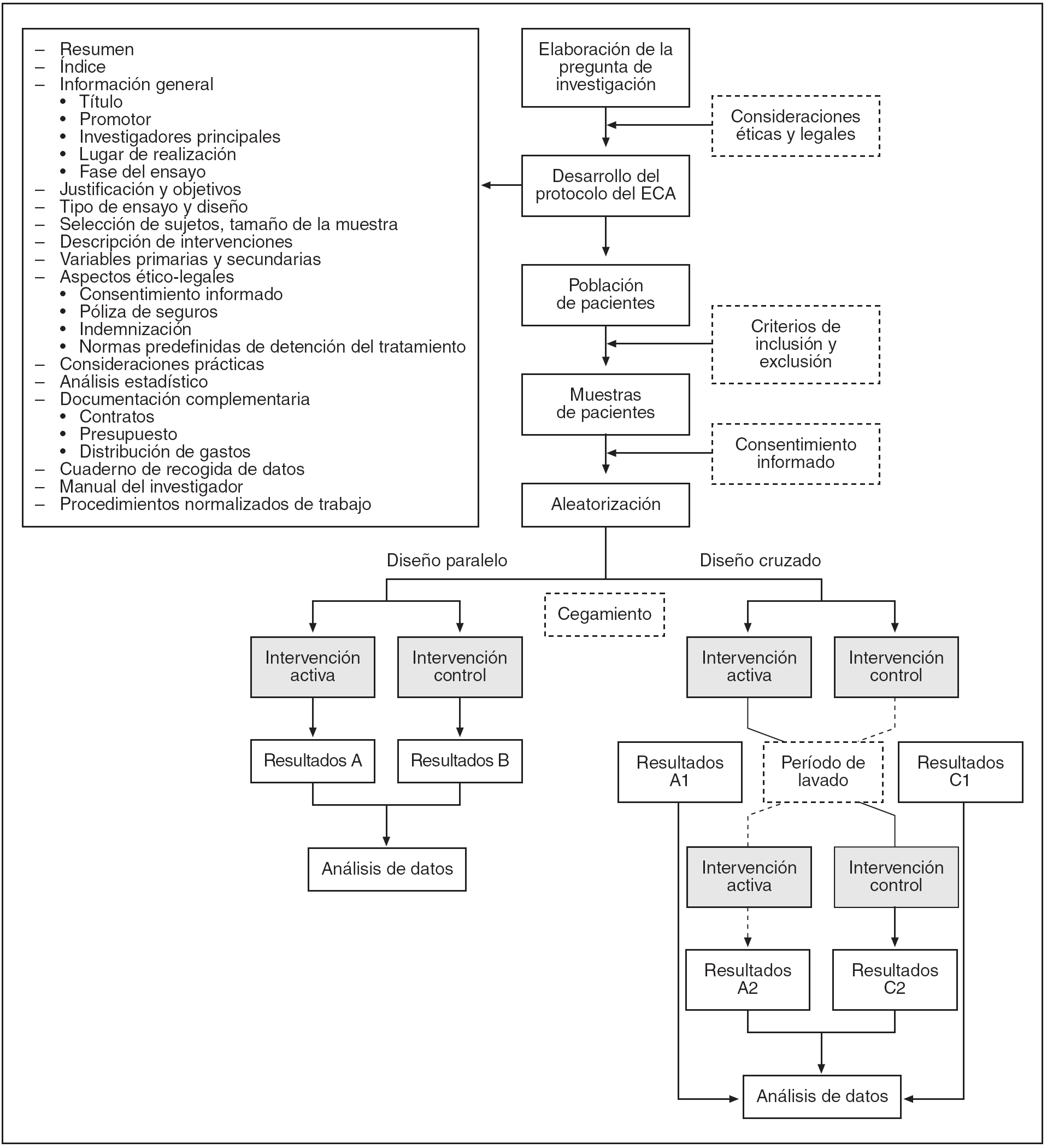
Figura 1. Etapas en la realización de un ensayo clínico aleatorio (ECA).
La determinación de un tamaño muestral adecuado pretende dotar a la muestra del poder estadístico suficiente para que si existen diferencias no debidas al azar entre los dos grupos sean detectadas. Para ello, además de tener en cuenta la homogeneidad de las poblaciones participantes, es necesario valorar la cuantía de las diferencias que se pretende poner de manifiesto, los errores de tipo I y II que se consideran aceptables y la proporción esperada de pacientes que abandonarán el estudio. En el capítulo siguiente de esta serie de investigación se tratará con más detalle cómo calcular tamaños muestrales.
Asignación aleatoria. Sesgo de selección
La aleatorización de los sujetos a los grupos de intervención define y diferencia el ECA de otros diseños epidemiológicos, como los estudios de cohortes. Consiste en la asignación de los sujetos participantes a los diferentes grupos de intervención por un mecanismo únicamente debido al azar, de modo que ni el investigador ni el sujeto pueden influir ni conocen el grupo al que son asignados1,4. Es la única herramienta metodológica que, teóricamente, permite una distribución equilibrada de las características de los pacientes a los grupos de tratamiento, generando grupos comparables respecto a cualquier condición conocida o no variables de confusión que pudiera afectar al desenlace de interés, de modo que la diferencia principal entre los grupos sea la intervención recibida. Es evidente que la eficacia de la aleatorización depende del tamaño muestral del estudio, por lo que en muestras pequeñas de sujetos, aunque sean adecuadamente aleatorizadas, existen más posibilidades de que la distribución de las características de los grupos de intervención no sea totalmente homogénea. Así, si tirásemos una moneda al aire 10 veces, con bastante probabilidad no nos saldrían exactamente 5 caras y 5 cruces; sin embargo, si la tiráramos 200 veces, la probabilidad de que la proporción de caras y cruces esté equilibrada, será mayor.
Existen diferentes métodos de aleatorización, aunque los más frecuentemente empleados son la aleatorización simple, aleatorización por bloques y aleatorización estratificada6. Un ejemplo de aleatorización simple, que es el procedimiento más intuitivo y fácil de aplicar, sería lanzar una moneda al aire, o de forma más sofisticada, el empleo de tablas de números aleatorios, o la generación de números aleatorios mediante un programa informático. Son sistemas sencillos, pero como se ha comentado, en muestras pequeñas no garantizan el equilibrio en el tamaño de los grupos, ni la distribución homogénea de los factores de confusión. Estos problemas se resuelven utilizando la aleatorización por bloques, que consiste en definir bloques de sujetos de un tamaño predeterminado en los que, por diseño, se fuerza a que el número de sujetos asignado a cada una de las intervenciones en estudio sea el mismo. Una vez constituidos los bloques se aleatorizan uno a uno los sujetos de cada bloque. Un último sistema frecuente para dividir a los sujetos en los grupos de intervención es la aleatorización estratificada. Para ello, se definen previmente los factores pronóstico por los que se desea estratificar (por ejemplo, sexo: hombre/mujer, y estado civil: soltero/casado/separado/viudo). Se constituyen tantos estratos como el producto del número de categorías de cada uno de los factores considerados (en nuestro ejemplo, dos por cuatro igual a ocho). Cada vez que se incorpora un nuevo sujeto al estudio se le asigna directamente a su estrato correspondiente (mujer soltera), y dentro de cada estrato se realiza un proceso de aleatorización independiente.
Además de la posibilidad de homogeneizar los grupos, evitando que sean otras variables diferentes de la intervención las que condicionen los resultados obtenidos, la aleatorización permite utilizar técnicas de enmascaramiento para evitar el denominado sesgo de selección7. Este sesgo tiene lugar cuando, o bien el investigador encargado de realizar la asignación, o bien el sujeto, tienen alguna influencia sobre el grupo al que será incorporado. No es difícil que el investigador conozca qué tratamiento corresponde a cada uno de los grupos, como es el caso de intervenciones que llevan asociados efectos adversos característicos. El interés personal del investigador en el éxito del nuevo tratamiento experimental puede influir en que trate de evitar que los sujetos con mejor pronóstico o mayores expectativas de mejoría vayan al grupo control, por ejemplo, aumentando el rigor a la hora de buscar criterios de exclusión por los que el sujeto no pueda participar en el estudio.
Para que ni las preferencias del investigador, ni las del paciente influyan en la decisión del grupo al que éste es asignado, es importante, lo primero, que la aleatorización se produzca cuando los sujetos ya estén incluidos en el estudio, una vez superados los criterios de selección y entregado el consentimiento informado de participación, y en segundo lugar, que la secuencia de asignación esté oculta para el investigador, ya que, al no conocer el tratamiento asociado a cada grupo su decisión no podrá influir en la inclusión de los sujetos a uno u otro. Este procedimiento se denomina "encubrimiento de la asignación" (del inglés, concealment allocation).
Exposición a las distintas intervenciones
Los ensayos clínicos se realizan fundamentalmente para evaluar tratamientos, ya sean farmacológicos, técnicas experimentales o intervenciones quirúrgicas, pero además, pueden realizarse para evaluar medidas preventivas, medidas de detección y diagnóstico de enfermedades, e incluso medios de mejora de la calidad de vida. En cualquiera de los casos, el estudio de una intervención debe realizarse conjuntamente con el de un grupo control, constituido por un conjunto de personas que comparte con el grupo activo todos los predictores de las variables de resultado, y se somete a lo largo del estudio a las mismas pautas y medidas, diferenciándose esencialmente en el tratamiento que reciben6.
De forma general, el grupo control de un ECA suele estar constituido por una de las siguientes opciones: un placebo, un tratamiento activo, o ninguna intervención. Los compuestos placebo son preparados que carecen de actividad farmacológica, pero cuyo aspecto y caracteres organolépticos son idénticos a los del preparado en estudio, y son diseñados para simular terapias médicas. También hablamos de técnicas placebo, constituidas por intervenciones o procedimientos que no tienen un efecto fisiológico ni bioquímico sobre la condición de estudio. La finalidad de emplear estos compuestos o procedimientos es controlar el denominado "efecto placebo", es decir, el efecto psicológico o fisiológico que tiene cualquier intervención, independientemente de su actividad, y que depende de la personalidad del paciente, del entusiasmo del equipo investigador, de las expectativas de mejoría, etc. Se produce una reducción de los síntomas, y en algunas ocasiones una aparición de efectos adversos, como resultado de la percepción de los pacientes de estar recibiendo una intervención terapéutica. Al comparar ambos grupos, es posible estimar el efecto real de la intervención estudiada, deduciéndolo del efecto logrado por el placebo en la misma situación clínica. De este modo, se puede estudiar la eficacia de una determinada intervención, ya que el placebo controla los efectos derivados de cualquier característica del tratamiento que no sea el efecto que se está estudiando. Si queremos estudiar, por ejemplo, la eficacia de un nuevo fármaco antihipertensivo, administrado vía oral en cápsulas rojas de un determinado tamaño, podemos emplear un grupo control constituido por cápsulas del mismo color rojo, tamaño, olor y sabor, pero formuladas con excipientes inactivos, carentes de actividad farmacológica alguna. Los sujetos, por un lado, no tendrán información sobre cuál es el tratamiento que están recibiendo (no es posible diferenciarlos), y por otro, tendrán las mismas expectativas de mejoría, misma susceptibilidad ante la aparición de eventos adversos, o las mismas reacciones fisiológicas ante la administración de un nuevo tratamiento.
Cuando existe una medida terapéutica aceptada como eficaz ante una determinada condición clínica, lo más adecuado para el estudio de una nueva intervención para tratar la misma condición es emplear la técnica conocida como intervención control. Utilizamos un tratamiento activo (el mejor tratamiento disponible) como grupo control, ya que, por un lado, no sería ético para los participantes emplear un placebo, y por otro, nos permite estimar la relación beneficio/riesgo del nuevo tratamiento ante una situación clínica concreta. Podemos, de este modo, acercarnos a la efectividad clínica del nuevo tratamiento, o realizar estudios de no inferioridad (demostramos que el nuevo tratamiento no es peor) y de equivalencia (demostramos que ambos son iguales) con intervenciones conocidas. Cualquier ventaja en cuanto a eficacia, seguridad, coste, adherencia al tratamiento, preferencia del paciente, etc. puede suponer un motivo para que el nuevo tratamiento constituya una alternativa terapéutica interesante ante esa misma dolencia.
Es importante, cuando comparamos dos tratamientos activos, desarrollar un método para cegar al paciente. Si, por ejemplo, quisiéramos comparar un nuevo antibiótico monodosis para el tratamiento de una determinada infección, con el que se emplea habitualmente, en dosis diarias durante cinco días, a los sujetos asignados al grupo de intervención, habría que administrarles el nuevo antibiótico el primer día, y los cuatro días restantes se les daría un preparado placebo para que continúen en el estudio a la par que los sujetos del grupo control, que estarían los cinco días recibiendo el fármaco de referencia. En este ejemplo, a igualdad de eficacia en los dos grupos, la pauta de administración puede constituir una ventaja importante ante el tratamiento convencional.
En algunos casos, por la propia estructura de la pregunta de investigación, lo más conveniente es emplear como grupo control sujetos sobre los que no se realiza ninguna intervención. Estos sujetos simplemente reciben los cuidados o atención que tienen habitualmente. Por ejemplo, para estudiar el efecto de una determinada intervención educativa en la prevención del hábito tabáquico en sujetos con riesgo cardiovascular elevado, podríamos emplear un grupo control constituido por sujetos sobre los que no se va a realizar ninguna intervención educativa, sino que simplemente se someterán a sus cuidados, revisiones y consejos dados por sus médicos y cuidadores habituales.
Es importante en la realización del ECA que respete el denominado principio de incertidumbre, según el cual sólo debe realizarse cuando existe verdadera incertidumbre sobre cuál de las intervenciones que se comparan beneficiará más al paciente. De lo contrario, si se realiza un ECA conociendo con certeza a priori que la intervención control es inferior a la experimental, no sería un comportamiento ético para los participantes del estudio, y además su publicación introduciría un importante sesgo en las evidencias disponibles sobre la eficacia de esa intervención, con las correspondientes implicaciones en la práctica de tales conclusiones.
Enmascaramiento (cegamiento). Sesgo de realización
Las técnicas de ciego, o de enmascaramiento, son aquellos procedimientos realizados con el fin de que algunos de los sujetos relacionados con el estudio no conozcan algunas características concretas de la realización de éste generalmente el tratamiento que recibirá cada sujeto, que pudieran ejercer un cambio en sus acciones o decisiones y sesgar los resultados6.
En función de los sujetos que han sido cegados a tal información se definen diferentes tipos de estudios1.
1) Ensayos abiertos son aquellos estudios en los que no se han utilizado técnicas de enmascaramiento. Todos los sujetos relacionados con el estudio conocen la intervención asignada a cada uno de los grupos.
2) Estudios simple ciego son aquellos en los que los sujetos participantes desconocen qué intervención están recibiendo.
3) Hablamos de doble ciego cuando tanto los pacientes como los investigadores desconocen el tratamiento administrado, y, por tanto, se elimina la posibilidad de que si el investigador conoce las intervenciones examine con más minuciosidad cualquier respuesta o posible efecto secundario experimentado por los sujetos. Existe, además, un triple ciego en los casos en los que el profesional estadístico que analizará los resultados tampoco conoce el tratamiento asignado a cada sujeto.
4) En los casos en los que no es posible el enmascaramiento de algunos de los sujetos implicados en el estudio (por ejemplo, no es posible enmascarar al investigador que aplica la técnica cuando ésta consiste en una intervención quirúrgica), o la variable respuesta es blanda e incluye elementos de subjetividad (percepción de mejoría, cuestionarios de síntomas, etc.), se emplea la técnica del evaluador ciego (evaluación ciega por terceros). En estos casos, la persona que ha de medir la variable respuesta no está involucrada directamente en la investigación, y desconoce el grupo al que pertenece cada uno de los sujetos, por lo que las mediciones e interpretaciones se realizarán de igual manera para todos los participantes, con independencia del tratamiento que hayan recibido.
El cegamiento permite que cualquier grado de imparcialidad sobre alguna de las intervenciones en estudio por parte del investigador, o bien del sujeto participante (autosugestión, confianza en una nueva intervención, etc.), pueda influir sobre los resultados o la interpretación de éstos. De este modo, se evita el denominado sesgo de realización7, que se da cuando existen diferencias en el trato o la atención sanitaria que reciben los sujetos en función del grupo al que pertenezcan. Si el grupo asignado es desconocido, todos los sujetos recibirán exactamente la misma atención, excepto la propia intervención.
El conocimiento de la intervención puede influir tanto en las actitudes del investigador que administra dicha intervención, como en el sujeto que la recibe, como en el profesional encargado de analizar los datos, introduciendo en cualquiera de los casos un importante sesgo en los resultados finales.
Aunque lo más deseable para la realización de un ECA es seguir una metodología, al menos, de doble ciego, ésta no siempre es posible. En el caso de intervenciones farmacológicas, los efectos secundarios o el perfil de toxicidad característico de algunos fármacos impide su cegamiento, así como en aquellos casos en los que no es posible disponer de una formulación galénica adecuada que actúe como grupo control placebo. Tampoco es posible el cegamiento cuando suponga riesgos innecesarios para el paciente, como en los casos en los que tendría que ser sometido a intervenciones quirúrgicas simuladas, o a administraciones repetidas de placebo por vía parenteral. Por último, en todos aquellos casos en los que por cualquier circunstancia el diseño doble ciego puede perjudicar la relación entre el médico y el paciente, no es conveniente llevarlo a cabo.
Descripción de pérdidas y abandonos. Sesgo de seguimiento
Otro aspecto metodológico a tener en cuenta en la realización de un ECA, es efectuar una correcta descripción de las pérdidas y abandonos experimentados durante su transcurso, es decir, recoger el número de sujetos que abandonan el estudio antes de su finalización, el grupo al que pertenecen y, si es posible, la causa del abandono o pérdida (aparición de evento adverso, falta de eficacia del tratamiento, etc.). Cada una de las causas tendrá un significado diferente en la valoración de la eficacia de la intervención en estudio, y, por tanto, disponer de esta información es muy importante para que no se produzca el denominado sesgo de seguimiento7.
A partir de esta información hay diferentes estrategias para analizar los datos y llegar a resultados válidos. Una de las opciones frecuentemente empleadas, es la realización de un análisis por intención de tratar. Según éste, cada paciente es analizado en el grupo al que fue asignado al inicio del estudio, independientemente de que no terminara la intervención que correspondía a ese grupo. Así, si realizáramos un ECA para estudiar la eficacia de dos intervenciones activas (A y B) con una duración de 5 días, y sobre una muestra de 100 sujetos, de los cuales 50 fueran aleatoriamente asignados al grupo A y 50 al grupo B, y durante el transcurso de esos 5 días hubieran abandonado el estudio 3 sujetos del grupo A y 7 del grupo B, un análisis por intención de tratar utilizaría los datos de los 50 sujetos iniciales del grupo A y los 50 del grupo B (no sólo 47 y 43 que fueron los que recibieron correctamente la intervención que les había sido asignada). Este tipo de análisis, además de permitir controlar el sesgo de seguimiento, presenta dos ventajas fundamentales. Por un lado, es la única estrategia que conserva las ventajas adquiridas mediante la asignación aleatoria de los sujetos, creando dos grupos comparables en todas las variables excepto en la intervención recibida, y además, es la estrategia que más se aproxima a la realidad de la práctica clínica diaria, donde los pacientes frecuentemente incumplen o rechazan los tratamientos que tienen prescritos.
REQUISITOS PARA REALIZAR UN ENSAYO CLÍNICO
Sujeto participante: derecho a la intimidad personal; consentimiento informado
Es necesario que todas las partes implicadas en un ECA guarden la más estricta confidencialidad para no vulnerar la intimidad personal ni familiar de los sujetos participantes en el mismo. Este derecho a la intimidad personal es una premisa establecida a priori para todos los participantes, y un derecho recogido tanto en la Constitución Española9, como en la Ley General de Sanidad10. Igualmente, es necesario tomar todas las medidas apropiadas para evitar el acceso de personas no autorizadas a los datos del ensayo.
Antes de ser incluido en un ECA, el sujeto deberá otorgar libremente su consentimiento informado, un procedimiento que garantiza que participará voluntariamente en el estudio después de haber comprendido la información que se le ha dado acerca de los objetivos de éste, beneficios, incomodidades y riesgos previstos, alternativas posibles, derechos y responsabilidades. Toda esta información debe ser facilitada al sujeto en términos que pueda comprender, y preferiblemente por escrito, dejándole la posibilidad de consultar cualquier duda acerca de la realización del estudio y de abandonarlo libremente en cualquier momento8. El resto de personas implicadas en el estudio evitarán cualquier influencia sobre el sujeto participante.
En la tabla 2 se recogen los datos que deben figurar tanto en una hoja de información para el posible participante, como en un modelo de consentimiento informado, de acuerdo con lo establecido en el Real Decreto 223/2004 de 6 de febrero, por el que se regulan los ensayos clínicos con medicamentos11.

Consideraciones éticas: informe del Comité Ético de Investigación Clínica
Ningún ECA podrá realizarse sin informe previo de un Comité Ético de Investigación Clínica. Los Comités de Ensayos Clínicos son los responsables de ponderar los aspectos metodológicos, éticos y legales del protocolo propuesto por el promotor del estudio. Su función, principalmente, es velar por los derechos de los sujetos sometidos voluntariamente a los ensayos clínicos, y suelen tener un carácter multidisciplinar, incluyendo componentes ajenos a las profesiones sanitarias. Es importante que conozcan y vigilen el cumplimiento de toda la normativa legal que rodea la realización de un ECA, con experimentación en humanos, desde los Principios de Buena Práctica Clínica y Garantía de Calidad y los principios éticos de la Declaración de Helsinki, hasta toda la normativa legal de cada comunidad autónoma, así como las disposiciones españolas y europeas elaboradas al respecto.
Básicamente, estudian los siguientes criterios5:
1) Evaluación de la idoneidad del protocolo en relación a los objetivos del estudio, eficiencia científica, posibilidad de alcanzar conclusiones válidas, justificación de los riesgos y molestias previsibles, ponderadas en función de los beneficios esperados para el sujeto y para la sociedad.
2) Evaluación de la idoneidad del equipo investigador para llevar a cabo el estudio propuesto: experiencia y capacidad investigadora, obligaciones asistenciales, o compromisos previamente establecidos con otros protocolos de investigación.
3) Evaluación de la información escrita que se facilitará a los posibles participantes y tipo de consentimiento que va a obtenerse.
4) Comprobación de la previsión de la compensación y tratamiento que se ofrecerá a los sujetos participantes en caso de lesión o daño atribuibles al ECA, y del seguro o indemnización para cubrir las responsabilidades especificadas por la legislación.
5) Conocimiento y evaluación del alcance de las compensaciones que se ofrecerán a los investigadores y a los sujetos de la investigación por su participación.
FORTALEZAS Y DEBILIDADES DEL ENSAYO CLÍNICO ALEATORIO
Debilidades
Los ECA son experimentos cuyo desarrollo lleva aparejada una gran complejidad. Desde el punto de vista del diseño, requieren un cierto dominio de la metodología para poder definir un protocolo adecuado a la población, a la intervención y a las medidas de resultado que queremos estudiar. Es preciso informar y organizar a los sujetos, que se enfrentarán a numerosas visitas, pruebas, tests, etc.. Por otro lado, las importantes restricciones éticas y legales para la realización de estos estudios hacen que no siempre sea posible someter a las personas a las condiciones ideales de experimentación.
Son estudios que sólo permiten evaluar el efecto de una única intervención y que suponen un elevado coste para llevarlos a cabo, el cual dependerá tanto de la duración del estudio como de la complejidad del protocolo establecido para su realización.
Otra de las principales debilidades del ECA es que son realizados en condiciones muy diferentes a las de su aplicación habitual. Tanto el proceso de selección de la muestra de estudio como las rígidas pautas que se siguen a la hora de realizar las intervenciones, convierten las condiciones del estudio en "excesivamente ideales" y alejadas de la realidad del resto de sujetos, por lo que los resultados obtenidos son, en ocasiones, difícilmente extrapolables y generalizables a la población.
Con respecto a los sujetos participantes se enfrentan a dos riesgos fundamentales. Por un lado, están recibiendo intervenciones cuya eficacia es, a priori, desconocida. Puede suceder que se trate de intervenciones completamente ineficaces o que hayan sido asignados a un grupo control constituido por una intervención placebo. De este modo, durante el período de duración del estudio no tienen las mismas posibilidades de prevenir o tratar su dolencia que los sujetos que sí estén recibiendo intervenciones de eficacia conocida. Al mismo tiempo, tampoco se tiene un conocimiento total del perfil de seguridad de la intervención estudiada, por lo que, aunque no suele ser habitual, podrían aparecer efectos secundarios graves desconocidos hasta entonces5.
Fortalezas
Los ECA son experimentos controlados. Es el investigador el que diseña un protocolo de actuación y decide cómo se llevará a cabo el estudio. Esto permite un mayor control del factor de estudio y, por tanto, definir mecanismos que cuiden de la seguridad del sujeto participante. Además son estudios prospectivos, por lo que el investigador participa y controla desde el comienzo, y es el que define su duración y el período de seguimiento postintervención durante el cual le conviene seguir a los participantes para obtener toda la información necesaria.
Los ECA permiten conocer y cuantificar la aparición de efectos secundarios indeseados consecuencia de la intervención en estudio. Con otros tipos de diseños, al no controlarse, o hacerlo en menor medida, los demás factores que podrían influir sobre estos efectos colaterales no es posible atribuirlos con certeza a la intervención estudiada.
Desde el punto de vista metodológico, por un lado, constituyen el diseño de investigación con mayor nivel de evidencia para comprobar hipótesis causales. La asignación aleatoria permite una distribución equitativa de todos los factores que pueden influir en el resultado, permitiendo aislar el efecto de la intervención del resto de factores y, por tanto, definiendo la causa real de los resultados obtenidos. Por otro lado, los ECA nos permiten un estudio de diferentes características de la intervención estudiada: eficacia, efectividad, eficiencia o equivalencia, en función del grupo control empleado o de las características de aplicación de dicha intervención.
Por último, desde el punto de vista del paciente, los ensayos clínicos suponen, de forma particular, la posibilidad de recibir tratamientos nuevos a los que otros pacientes no tienen acceso, y que, en algunos casos, constituyen la última alternativa disponible ante una determinada dolencia12. Estos pacientes recibirán una elevada y especial atención médica durante toda la duración del estudio. De forma general, los ECA constituyen la mejor posibilidad de crear nuevas alternativas terapéuticas y de poner en el mercado, y por tanto al alcance del paciente, intervenciones preventivas, terapéuticas o rehabilitadoras que constituyen nuevas soluciones para la práctica asistencial diaria, con expectativas de mejorar la calidad de vida y el estado de salud del individuo.
Correspondencia: J.L. R. Martín.
Área de Investigación Clínica.
Fundación para la Investigación Sanitaria en Castilla la Mancha (FISCAM). Edificio Bulevar.
C/ Berna, n.º 2, local 0-2.
45003 Toledo.
Correo electrónico: jlrmartin@jccm.es
Recibido el 22-05-07; aceptado para su publicación el 22-05-07
