




El sistema hemostático, constituido por componentes celulares y proteínas plasmáticas solubles, mantiene la sangre en estado fluido1. En respuesta a una lesión endotelial, las plaquetas se adhieren al subendotelio expuesto, se activan, agregan otras plaquetas circulantes y proporcionan la superficie fosfolipídica adecuada para el ensamblaje de diferentes compuestos enzimáticos de la cascada de la coagulación que generan gran cantidad de trombina en esa zona.
El sistema de la coagulación
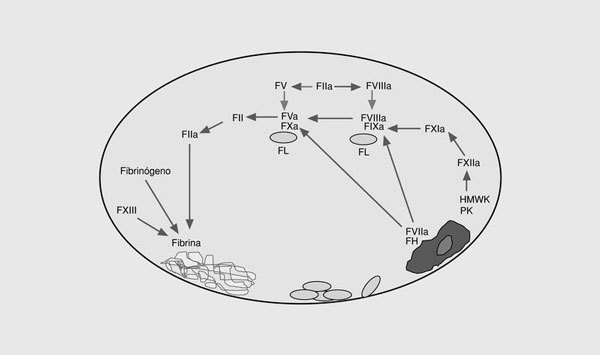
La coagulación sanguínea es el conjunto de reacciones que dan lugar a la formación de trombina, enzima clave de la coagulación, en el punto de lesión vascular (fig. 1)2. Este sistema se inicia en la superficie de las células endoteliales a través de la exposición del factor tisular al torrente sanguíneo. Éste se une al factor VII activado (FVIIa), y el complejo enzimático resultante activa los factores IX y X3. Los FVIIa, FIXa y FXa intervienen en un sistema de retroalimentación positiva al activar el factor VII unido al factor tisular. Por otra parte, el FIXa por la vía del factor tisular, activa el factor X, en una reacción que es acelerada por un cofactor, el FVIIIa. La exposición del factor tisular da lugar a la vía extrínseca de la coagulación y es el mecanismo por el cual se inicia la coagulación in vivo en respuesta a la lesión vascular. Otra vía de la coagulación es la vía intrínseca formada por el FXII, el quininógeno de alto peso molecular, la precalicreína y el FXI. El papel fisiopatológico de esta vía no está completamente aclarado ya que no interviene significativamente en la coagulación a partir de la lesión vascular y, además, los déficit congénitos de las proteínas de este sistema no provocan problemas hemorrágicos excepto la deficiencia del FXI. El resultado final de las dos vías descritas es la activación del FX. El FXa forma con su cofactor, el FV, el complejo protrombinasa que convierte el FII (protrombina) en FIIa (trombina). La activación del factor V se produce por el FXa y la trombina. En la etapa final de la coagulación, la trombina divide el fibrinógeno para generar monómeros de fibrina que se polimerizan uniéndose unos a otros para formar un coágulo que es estabilizado por el FXIII. La trombina también se retroalimenta activando al FVIII y al FIX. El FVIII circula unido al factor von Willebrand4 y, una vez activado, se separa de éste y forma junto al FIXa en la superficie plaquetaria un complejo que activa al FX. La activación del FXI por la trombina es otro sistema de retroalimentación que da lugar a la generación de FIXa, el cual activa a su vez al FX5.
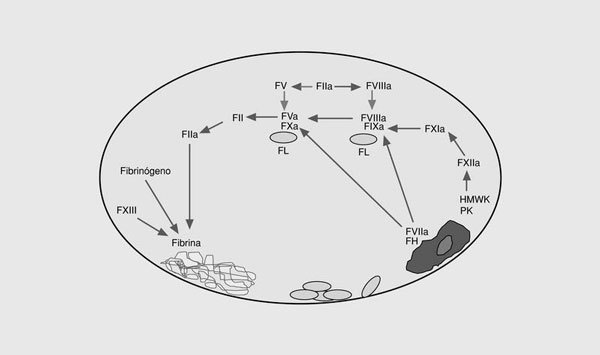
Fig. 1. Esquema de las vías intrínseca y extrínseca de la coagulación. FT: factor tisular; HMWK: kininógeno de alto peso molecular; PK: precalicreína; FL: fosfolípidos.
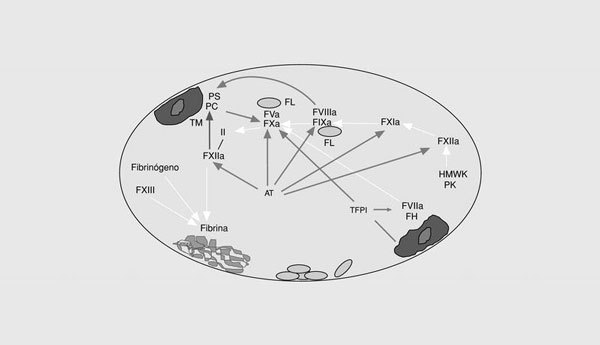
La cascada de la coagulación sanguínea tiene la capacidad de amplificar un pequeño estímulo inicial para poder formar un coágulo de fibrina. Sin embargo, la naturaleza explosiva de este sistema no puede existir sin mecanismos de regulación (o anticoagulantes naturales) que eviten una coagulación masiva (fig. 2). Así, el inhibidor de la vía del factor tisular (TFPI) es una proteína plasmática asociada a una lipoproteína que forma un complejo cuaternario con el factor tisular, el FXa y el FVIIa e inhibe la vía extrínseca de la coagulación6. Muchos de los factores activados durante la activación de la coagulación son inhibidos por la antitrombina. Se trata de una proteína que inhibe la actividad de las enzimas de la vía intrínseca y común de la coagulación. En presencia del heparán sulfato endógeno, la tasa de inactivación se incrementa 200 veces más. En presencia de trombomodulina unida a las células endoteliales, la trombina activa la proteína C, que, a su vez, destruye el FVa y el FVIIIa7. Como otras reacciones de la hemostasia, la acción de la proteína C activada es acelerada por un cofactor, en este caso, la proteína S. Finalmente, diferentes reacciones del sistema de la fibrinólisis culminan con la formación de la fibrina, una serinproteasa que elimina el coágulo de fibrina del vaso. Por tanto, el correcto funcionamiento del sistema hemostático depende del adecuado balance entre las reacciones procoagulantes, por un lado, y las anticoagulantes y fibrinolíticas, por otro. Cualquier anomalía que afecte a este sistema puede alterar dicho equilibrio y ocasionar estados de riesgo trombótico o hemorrágico.
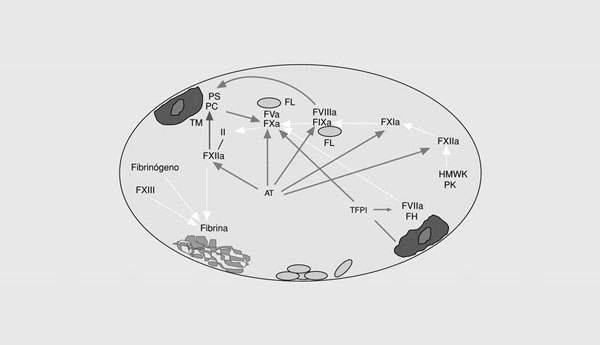
Fig. 2. Esquema de los sistemas anticoagulantes naturales de la coagulación. TFPI: inhibidor de la vía del factor tisular; AT: antitrombina; PC: proteína C; PS: proteína S; TM: trombomodulina.
El sistema de la fibrinólisis
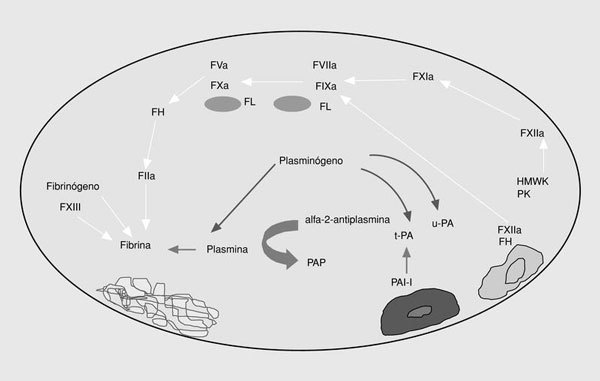
El sistema de la fibrinólisis es una cascada enzimática que consta de una serie de activadores e inhibidores que regulan la conversión del plasminógeno en plasmina. La generación de plasmina libre en la superficie del trombo conduce a la lisis de la fibrina, dando lugar a los productos de degradación de la fibrina (fig. 3).
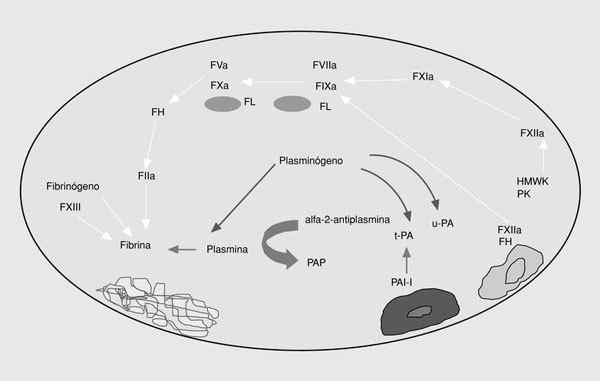
Fig. 3. Esquema de la fibrinólisis. t-PA: activador tisular del plasminógeno; PAI-1: inhibidor tipo 1 del t-PA; u-PA: urocinasa; PAP: complejos plasmina-antiplasmina.
La regulación del sistema de la fibrinólisis está mediada por interacciones moleculares específicas entre sus principales componentes y por la síntesis y pos terior liberación a partir de las células endoteliales de los activadores e inhibidores de los activadores del plasminógeno. Por tanto, un incremento de la actividad del sistema de la fibrinólisis favorece la aparición de trastornos hemorrágicos, mientras que el defecto de la actividad fibrinolítica puede predisponer a la trombosis.
Componentes del sistema de la fibrinólisis
Las enzimas del sistema de la fibrinólisis son proteasas del tipo serina, es decir, su locus activo está compuesto por los aminoácidos serina, ácido aspártico e histidina, dando lugar a la llamada región catalítica. Este locus activo se localiza en la región carboxiterminal de las moléculas, mientras que las regiones aminoterminales contienen uno o más dominios estructurales y funcionales, como los dominios finger (por analogía con los finger de la fibronectina), el dominio factor de crecimiento epidérmico (EFG) y los dominios kringle. Los inhibidores del sistema de la fibrinólisis son miembros de la superfamilia de las serpinas (inhibidores de las serinproteasas). En este caso, en el extremo carboxiterminal poseen un péptido reactivo específico (arginina-X o lisina-X) que está unido a su enzima. Ello da lugar a una estructura inactiva con una función de inhibición enzimática. Las características químicas y genéticas de los principales componentes del sistema de la fibrinólisis quedan resumidas en la tabla 1.

Plasminógeno
El plasminógeno humano es una glucoproteína de cadena simple de 92 kD formada por 791 aminoácidos y 24 puentes disulfuro8. La molécula se organiza en siete dominios estructurales, repartidos en un «péptido de preactivación» (aminoácidos 1-77), cinco dominios secuenciales homólogos, denominados kringle (estructuras de tres bucles unidos por puentes disulfuro de 80 aminoácidos cada una de ellas), y el dominio proteasa (aminoácidos 562-791). Los dominio kringle contienen lisina en los puntos específicos que median la unión del plasminógeno a la fibrina y la interacción de la plasmina con la α2-antiplasmina. Por tanto, desempeñan un papel crucial en el reconocimiento de la fibrina, de la superficie celular y de la α2-antiplasmina. El plasminógeno se convierte en plasmina al romperse la unión Arg561-Val562. El gen del plasminógeno está localizado en el brazo largo del cromosoma 6, concretamente en las bandas q26 o q27, y consta de 19 exones y 18 intrones9. Cada uno de los cinco dominios kringle es codificado por dos exones separados por un intrón situado en el centro de cada estructura.
Activador tisular del plasminógeno
El activador tisular del plasminógeno (t-PA) es una proteasa serínica de 68 kD, compuesta por 530 aminoácidos10. La molécula está organizada en varios dominios como el dominio finger, que comprende los aminoácidos 4-50, el dominio EFG, con los aminoácidos 50-87, y dos dominios kringle, con los aminoácidos 87-176 y 176-262, que se localizan en el extremo aminoterminal de la molécula. Por el contrario, en el extremo carboxiterminal se localiza el dominio de la proteasa que comprende los aminoácidos 276-527, constituyendo la llamada región catalítica10. Estos diferentes dominios median diferentes funciones de la enzima. Así, la unión del t-PA a la fibrina está mediada fundamentalmente por el dominio finger y el segundo de los dominios kringle. Por otra parte, el t-PA es degradado por la plasmina, por hidrólisis del puente Arg275-Ile276, a una estructura de dos cadenas inactiva. El gen del t-PA se localiza en el cromosoma 8 (bandas 8.p.12-q.11.2) y está compuesto por 14 exones y 13 intrones11.
Urocinasa
El scu-PA o prourocinasa es una glucoproteína de 54 kD que contiene 411 aminoácidos12. En este caso, la tríada catalítica se localiza igualmente en el extremo carboxiterminal, mientras que en el aminoterminal se localizan el dominio EFG y un dominio kringle. El dominio EFG es el responsable de la unión del scu-PA a su receptor, el cual está presente en la superficie de varios tipos celulares. La scu-PA se transforma en tcu-PA o urocinasa por la rotura de la unión Lis158-Ile159. El gen de la tcu-PA se localiza en el cromosoma 10. Contiene 11 exones y la organización intrón-exón del gen se asemeja bastante a la del gen del t-PA11.
Receptor del activador del plasminógeno tipo urocinasa
El receptor del activador del plasminógeno tipo urocinasa (u-PAR) es una glucoproteína de 50-60 kD compuesta por 313 aminoácidos que se une a la tcu-PA a través de los dominios EFG de ésta. Está formado por tres dominios estructurales homólogos, de los cuales el fragmento aminoterminal es el que se une a la tcu-PA13. La unión de scu-PA al u-PAR parece ser crucial para la activación de la tcu-PA. Esta unión provoca un aumento en la generación de plasmina debido, por una parte, a la activación del plasminógeno y, por otra parte, a la activación por retroalimentación de scu-PA en tcu-PA por la plasmina generada. El gen del u-PAR se localiza en el cromosoma 19, en las bandas q13.1-q13.2, y está constituido por 7 exones14.
Alfa-2-antiplasmina
La molécula de α2-antiplasmina fue descrita originalmente como una glucoproteína de 452 aminoácidos. Actualmente, se sabe que en realidad son 464 aminoácidos los que forman la estructura de esta glucoproteína de 70 kD15. Posee una característica única entre las serpinas, ya que su extremo carboxiterminal de 51 aminoácidos contiene un locus secundario de unión que reacciona con los sitios de unión de lisina de los dominios kringle 1-3 del plasminógeno y de la plasmina16. El gen de la α2-antiplasmina está localizado en el cromosoma 18, bandas p11.1-q11.2. Contiene 10 exones, y el exón IV es el encargado de codificar la región aminoterminal donde se encuentra el punto de unión de la fibrina. El exón X, por su parte, es el que codifica la región carboxiterminal en donde se sitúa el lugar de unión para el plasminógeno.
Inhibidor del activador tisular del plasminógeno
Los dos principales inhibidores del activador tisular del plasminógeno (PAI) son el PAI-1 y el PAI-2. El PAI-1 es una glucoproteína de cadena única de 52 kD compuesta por 379 aminoácidos17. La molécula de PAI-1 se estabiliza a través de su unión con la proteína S o vitronectina. El gen del PAI-1 se localiza en el cromosoma 7, bandas q21.3-q22, y está constituido por 9 exones. El PAI-2 es una serpina de 393 aminoácidos18, de la que existen dos formas diferentes con propiedades cinéticas similares; una forma intracelular no glucosilada de 47 kDa y una forma glucosilada de 60 kDa. La función de PAI-2 intracelular es desconocida ya que su enzi ma de unión, la tcu-PA, es extracelular. Se cree que podría comportarse como un pool del cual el PAI-2 podría secretarse en caso de lesión celular19. El gen del PAI-2 se localiza en el cromosoma 18, bandas q21-q23. Contiene 8 exones y su estructura varía ligeramente del del PAI-1.
Activación del plasminógeno a plasmina
Todos los activadores del plasminógeno convierten a éste en plasmina a través de la rotura de la unión Arg561-Val562. La molécula de plasmina que se forma es de doble cadena y está compuesta por una cadena pesada que contiene los cinco kringles (extremo aminoterminal del plasminógeno) y una cadena ligera (extremo carboxiterminal) que contiene la región catalítica, compuesta por His603, Asp646 y Ser741.
Inhibición de la plasmina por α2-antiplasmina
La α2-antiplasmina forma, junto a la plasmina, un complejo 1:1 inactivo. Esta inhibición se lleva a cabo mediante dos reacciones consecutivas: la primera, rápida, da lugar a un complejo inactivo reversible que es seguida por una segunda reacción, más lenta, de la que resulta un complejo inactivo irreversible. La vida media de las moléculas de plasmina generadas en la superficie de la fibrina es 2 o 3 veces mayor que la de la fibrina libre o circulante20.
Mecanismo de acción del activador tisular del plasminógeno
En presencia de fibrina
El t-PA es una enzima con poca actividad en ausencia de fibrina. Sin embargo, en presencia de ésta aumenta de manera notable el grado de activación sobre el plasminógeno21. La formación de los monómeros de fibrina mediada a partir de la trombina y su posterior polimerización son esenciales para la estimulación del plasminógeno a partir del t-PA. El grado óptimo de estimulación sólo se consigue después de la rotura prematura de la cadena Aα del extremo carboxiterminal y de la cadena Bβ del fragmento aminoterminal de la fibrina, lo que da lugar a los polímeros X22. Existen datos que sugieren que la fibrina actúa como la superficie en la cual el t-PA y el plasminógeno formarían un complejo ternario. La formación de este complejo da lugar a un aumento de la afinidad del t-PA por el plasminógeno23.
Recientemente, se ha caracterizado el inhibidor de la fibrinólisis activable por la trombina (TAFI), el cual se activa por el complejo trombina-trombomodulina24. Se trata de una carboxipeptidasa que inhibe la fibrinólisis, eliminando residuos Arg o Lys en el extremo carboxiterminal de la fibrina parcialmente degradada. Con ello inhibe la lisis de la fibrina por la plasmina activada por el t-PA.
En la superficie celular
Algunos tipos celulares poseen la capacidad de unir a los activadores del plasminógeno y al mismo plasminógeno en su superficie, lo que da lugar a un aumento de la activación de éste25 y, a la vez, protege a la plasmina unida de su inhibición por la α2-antiplasmina26. Algunos estudios han demostrado que el endotelio regula la fibrinólisis pericelular al modular la expresión de los receptores del plasminógeno27. Una proteína de membrana de 40 kD (relacionada con la anexina II) se ha propuesto como el receptor funcional del t-PA28, de manera que el t-PA unido a la superficie celular mantiene su actividad enzimática y, además, queda protegido de la inhibición del PAI-1. Por tanto, la unión del plasminógeno y de sus activadores al endotelio vascular favorece la generación de plasmina y puede ejercer un papel importante en el mantenimiento del fluido sanguíneo.
Estos receptores celulares también pueden intervenir en la eliminación rápida del t-PA del torrente circulatorio. Así, las células endoteliales hepáticas poseen unos receptores que reconocen los kringle 1 y las células del parénquima hepático contienen un receptor dependiente del calcio que es capaz de interaccionar con los dominios finger y/o EFG del t-PA. Los hepatocitos contienen, además, receptores de alta afinidad para la unión y degradación de los complejos t-PA/PAI siendo también capaces de unirse, aunque con menor afinidad, al t-PA circulante.
Mecanismo de acción de la urocinasa
En presencia de fibrina
En el plasma, en ausencia de fibrina, la scu-PA es estable y no activa el plasminógeno. En presencia del coágulo de fibrina la scu-PA induce la lisis específica de este coágulo29. Además, la α2-antiplasmina impide la conversión de scu-PA a tcu-PA fuera del coágulo y mantiene su especificidad por la fibrina. El fragmento E-2 de la fibrina estimula específicamente la activación del plasminógeno por la scu-PA. Scu-PA es, por tanto, un activador deficiente del plasminógeno cuando éste se presenta unido a los residuos de lisina de la fibrina intacta, pero presenta una alta actividad por el plasminógeno unido a residuos de lisina del extremo carboxiterminal formados a partir de la fibrina parcialmente degradada.
En la superficie celular
La unión de la scu-PA al u-PAR desempeña un papel crucial en su activación en condiciones fisiológicas. Como se ha mencionado anteriormente, esta unión provoca un aumento en la generación de plasmina debido, por una parte, a la activación del plasminógeno y por otra parte, a la activación por retroalimentación de scu-PA en tcu-PA por la plasmina generada30. La plasmina unida a la célula queda protegida de su inactivación por la α2-antiplasmina; además, favorece la activación de la scu-PA unida a su receptor. Este sistema se puede inhibir eficazmente por el PAI-1 y el PAI-231. Si bien se ha sugerido que la activación del plasminógeno se produciría a partir de la formación de un complejo que dependería del u-PAR, los mecanismos por los que funciona este receptor son, hoy por hoy, desconocidos.
Inhibición de los activadores del plasminógeno
En la inhibición del t-PA humano se han involucrado numerosos mecanismos. Así, el PAI-1 es un inhibidor rápido del t-PA que, en condiciones normales, se encuentra en el plasma a bajas concentraciones. Además, el t-PA se inhibe de forma más lenta por la α2-antiplasmina, α1-antitripsina y el C1-inhibidor. Sin embargo, la principal vía de eliminación del t-PA del torrente circulatorio es la hepática.
En el plasma humano, la tcu-PA es lentamente inhibida por varias proteasas, como α2-macroglobulina, α1-antitripsina, antitrombina, α2-antiplasmina y el PAI-3, que corresponde al inhibidor de la proteína C activada32, aunque el PAI-1 y el PAI-2 son responsables de su inhibición de forma más rápida y específica. A diferencia de la tcu-PA, la scu-PA no se inhibe por proteasas plasmáticas sino que el principal mecanismo de eliminación es la vía hepática.
El PAI-1 reacciona con el t-PA de cadena simple y de doble cadena y con la tcu-PA pero no con la scu-PA. Las regiones de la molécula de t-PA y de tcu-PA con carga positiva son las responsables de esta rápida reacción. El PAI-1 se elimina del torrente circulatorio a través de la vía hepática33. Por su parte, el PAI-2 inhibe más lentamente la tcu-PA que el PAI-1. También actúa de forma eficaz sobre el t-PA de doble cadena, de forma menos eficaz sobre el t-PA de cadena simple y no inhibe la scu-PA.
Síntesis y secreción de los activadores del plasminógeno
Las células endoteliales sintetizan y secretan el t-PA al torrente circulatorio. Se ha demostrado que estas células sintetizan más t-PA si existe lesión arterial y que la estimulación del endotelio vascular en forma de oclusión venosa, infusión de DDAVP o epinefrina y el ejercicio físico provocan una rápida liberación (en minutos) de t-PA34. Esta respuesta es demasiado rápida para ser explicada con un aumento de la síntesis de t-PA y, por tanto, puede reflejar la liberación de los depósitos celulares de t-PA, aunque la existencia de éstos no ha sido confirmada. Algunas sustancias como la trombina que estimula la liberación de t-PA de las células endoteliales también estimula la secreción de PAI-1. La síntesis de t-PA por las células endoteliales también se incrementa por una gran variedad de sustancias como la trombina, la histamina, del factor de crecimiento fibroblástico y la proteína C activada. Sustancias vasoactivas como la histamina y la trombina, a través de la activación de la fosfolipasa C a través de ésta, activando la proteinquinasa C regulan la síntesis de t-PA. Mientras esta síntesis de t-PA tiene lugar fundamentalmente en las células endoteliales, la de scu-PA se produce en otras variedades celulares del organismo, como fibroblastos, células epiteliales y neumocitos.
Síntesis y secreción de los inhibidores de los activadores del plasminógeno
La existencia de ARNm de PAI-1 en una gran variedad de tejidos sugiere que numerosos y diversos tipos celulares, como el endotelio o las células de músculo liso, pueden ser su lugar de producción. El PAI-1 se localiza en el plasma, las plaquetas, la placenta y la matriz extracelular. Excepto en las plaquetas, que contienen esencialmente PAI-1 inactivo, el PAI-1 no se almacena en las células y sí, en cambio, se secreta rápidamente después de su síntesis, de forma que posee un ritmo circadiano con una concentración plasmática más elevada por la mañana y más baja por la tarde y por la noche, mientras que el t-PA presenta una variación circadiana opuesta.
La síntesis y secreción de PAI-1 está modulada por varios agonistas, como hormonas, factores de crecimiento, endotoxinas y citocinas. En las células endoteliales, la expresión del gen del PAI-1 se estimula por los lipopolisacáridos, la interleucina 1, el factor de necrosis tumoral alfa, el factor de crecimiento fibroblástico, la trombina, las lipoproteínas de muy baja densidad y la insulina. Por otra parte, en las células endoteliales adyacentes al coágulo, en las células del músculo liso de la neoíntima y en los macrófagos, la expresión de ARNm está aumentada, detectándose PAI-1. Este aumento de su expresión en la pared arterial inducida por la trombosis puede provocar un desequilibrio entre la fibrinólisis y la trombosis a favor de esta última35.
El PAI-2 se ha identificado en la placenta y en el plasma de embarazadas y se secreta por los leucocitos y por las células de fibrosarcoma. Su secreción se activa por la endotoxina que estimula la transcripción del gen del PAI-236.
Factores de riesgo trombótico
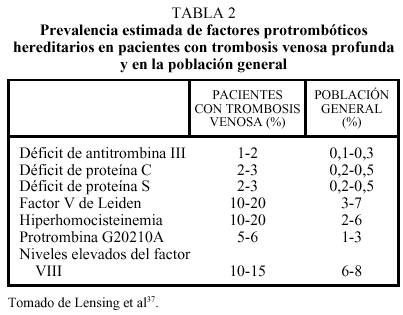
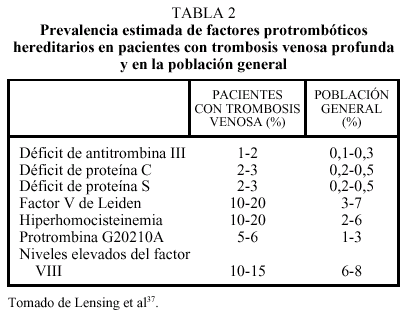
Los llamados estados hipercoagulables congénitos están constituidos por anomalías protrombóticas bien establecidas, la mayoría de las cuales son hereditarias. Tradicionalmente, estas causas genéticas de trombofilia se han estudiado en el contexto de familias con tendencia a padecer trombosis. La prevalencia estimada de estas alteraciones en la población general es de 1 cada 3.000-5.000 habitantes, mientras que una o más de estas anomalías trombofílicas pueden encontrarse en un 40-60% de los pacientes con un primer episodio de trombosis venosa37. Sin embargo, la prevalencia de estos factores en pacientes con trombosis es muy variable y oscila entre el 20% del factor V de Leiden y el 1% del déficit de antitrombina (tabla 2)37. Finalmente, se están evaluando en la actualidad nuevas anomalías, entre las que destacan un gran número de polimorfismos (de los factores II, VII, XII y XIII, de las proteínas C y S, del t-PA y tcu-PA y del PAI-1)38, de los que quizás los más prometedores sean los valores plasmáticos de FVIII y FXI. En dos situaciones pueden coexistir fenómenos trombóticos arteriales y venosos: el síndrome antifosfolipídico relacionado con la presencia de anticuerpos dirigidos contra complejos de fosfolípidos y proteínas y la hiperhomocisteinemia.
Existen, en segundo lugar, los denominados estados hipercoagulables adquiridos constituidos por un grupo heterogéneo de procesos en los que existe un riesgo elevado de aparición de trombosis cuando se compara con el de la población general. Entre ellos destacan el embarazo, la edad avanzada, la inmovilización, el uso de estrógenos, la cirugía, las neoplasias, el síndrome nefrótico, los síndromes mieloproliferativos y la hemoglobinuria paroxística nocturna, los cuales pueden cursar con trombosis arteriales y venosas39.
Déficit de antitrombina
Se transmite de forma autosómica dominante y su prevalencia en la población general varía entre el 0,02% en donantes sanos40 y el 0,5-1%41 en una población de pacientes con trombosis no seleccionados. Se transmite de forma autosómica dominante y la forma homozigota es incompatible con la vida. Característicamente, los pacientes afectados desarrollan trombosis antes de los 25 años42. Es la alteración en la que la probabilidad de desarrollar trombosis en individuos afectados es más elevada. Menos del 1% de todos los fenómenos trombóticos se deben a este déficit43.
Déficit de proteína C
La deficiencia heterozigota de proteína C se ha asociado a un aumento del riesgo trombótico44. Se transmite de forma autosómica dominante y su forma homozigota ocasiona graves trombosis neonatales. La prevalencia de trombosis en estos pacientes es del 3%41,45 y su prevalencia en la población general es del 0,2%46. Todo ello hace que el riesgo relativo de trombosis asociado a este déficit de proteína C sea de 1543. Entre el 1 y el 2% de todos los fenómenos trombóticos se debe a este déficit32.
Déficit de proteína S
Desde 1984 se han descrito familias con este déficit47, de manera que su herencia es autosómica dominante y su frecuencia es similar al déficit de proteína C. El mismo defecto genotípico puede dar lugar a variables fenotípicas como el tipo I (proteína S total baja y proteína S libre baja) o el tipo III (proteína S libre baja con proteína S total normal)48. Si a esto unimos el efecto de las variables adquiridas (sobre todo la edad), el espectro clínico de trombosis en este déficit es muy amplio48. Se ha comunicado que el 1% de todos los fenómenos trombóticos se deben a este déficit43.
Resistencia a la proteína C activada y factor V de Leiden
En 1993, Dahlbäck et al49 describieron tres pacientes con trombosis que eran portadores de un nuevo defecto en la vía anticoagulante de la proteína C en forma de una disminución de la actividad anticoagulante del plasma frente a la proteína C activada o resistencia a la proteína C activada (RCPA). Un año más tarde, el mismo autor50 y Bertina51 llegaron a la conclusión de que el 80% de todos los individuos con RPCA eran portadores de una mutación en el exón 10 del gen del factor V, constituyendo el llamado factor V de Leiden. La mutación consiste en un cambio G por A en la posición 1.691 del gen que codifica el factor V51, de forma que en la población caucásica constituye la mutación genética que da lugar a trombosis más prevalente52-54. Se presenta en el 20% de los pacientes con trombosis55 y es, por tanto, la anormalidad genética más frecuente en pacientes con trombosis. Su prevalencia en la población sana de origen caucásico varía entre el 2 y el 15%56. La presencia de la mutación está asociada a un riesgo incrementado de trombosis de 3 a 8 veces en la forma heterozigota y de al menos 80 veces en la forma homozigota53.
El estado de RPCA también puede aparecer en ausencia del factor V de Leiden. En este caso puede ser secundaria a causas de origen genético o adquiridas. Entre las primeras destaca la reciente descripción de otra mutación en el factor V (factor V de Cambridge), que afecta a otro punto de acción de la proteína C sobre el factor V, mientras que el embarazo, los anticonceptivos orales o la existencia de un anticoagulante lúpico son causas adquiridas de RPCA.
Mutación G20210A del gen de la protrombina
Descrita en 1996, esta mutación constituye la segunda situación de riesgo trombótico más prevalente y está presente en el 6% de los pacientes con trombosis 58 y se ha descrito un aumento del riesgo trombótico de 2 a 5 veces en relación con su presencia 59.
Hiperhomocisteinemia
Los valores plasmáticos elevados de homocisteína se asocian con un aumento del riesgo de trombosis arteriales y, de forma dudosa, de trombosis venosas60. La hiperhomocisteinemia puede ser consecuencia de factores adquiridos o genéticos61 de manera que la mayoría de los individuos con un aumento de la misma no son portadores de ninguna variación genética, sino que presentan un deterioro del metabolismo de la metionina, en el que la hiperhomocisteinemia está producida por un consumo alimentario insuficiente de ácido fólico y vitaminas B6 o B1262. Entre las variaciones genéticas destacan las mutaciones de la cistationina β-sintetasa o de la metilén-tetrahidrofolato reductasa63. Un polimorfismo frecuente de esta última, el C677T, se asocia con la presencia de una enzima termolábil y valores elevados de homocisteína64, aunque su papel como factor de riesgo trombótico es controvertido64.
Anticuerpos antifosfolipídicos
Los anticuerpos antifosfolipídicos, ya sea en forma de anticoagulante lúpico o anticuerpos anticardiolipina, constituyen un factor de riesgo trombótico adquirido que se relaciona con el desarrollo tanto de trombosis arteriales como venosas. Junto a la trombocitopenia y a los abortos de repetición, constituyen el llamado síndrome antifosfolipídico65. Actualmente, se sabe que, en realidad, estos anticuerpos están dirigidos contra un complejo de fosfolípido y proteína (o cofactor), de las cuales se han reconocido la β2-glucoproteína I66 y la protrombina67. La presencia de anticoagulante lúpico estaría presente entre el 2 y el 14% del total de las trombosis43.
Anomalías congénitas del fibrinógeno
Aproximadamente un 12% de las denominadas disfibrinogenemias presentan episodios trombóticos, de manera que se han descrito cuadros de trombosis graves, venosas o arteriales en 34 familias, mientras que otras 6 variantes presentaban simultáneamente hemorragias y trombosis68. Los mecanismos descritos en estas anomalías consisten en un aumento de la resistencia de la fibrina a la lisis por la plasmina o bien un defecto de la activación del plasminógeno por el t-PA en presencia de fibrina.
Anomalías congénitas del plasminógeno
En 1978 se detectó la existencia de una displasminogenemia en una familia japonesa con tendencia trombótica grave. Desde entonces se han descrito diversas anomalías moleculares del plasminógeno, con alteración en el centro activo y activación defectuosa a plasmina, asociadas a trombosis venosas recurrentes69. En la mayoría de casos en los que se conoce el defecto molecular, éste consiste en una sustitución en el lugar activo del amoninoácido lisina en posición 600 por treonina. En otros casos se trata de moléculas de plasminógeno con menor afinidad por sus activadores.
Valores elevados de factor VIII
El FVIII actúa como un reactante de fase aguda y su origen es sólo parcialmente conocido. En aquellos pacientes que presenten valores plasmáticos persistentemente elevados a lo largo del tiempo puede existir una predisposición genética. La prevalencia de valores elevados de este factor en pacientes con trombosis venosa es de aproximadamente el 15%70, por lo que en el futuro su determinación se incluirá, muy probablemente, en el cribado de rutina de trombofilia.
Valores elevados de factor XI
El déficit de FXI se asocia clínicamente a manifestaciones hemorrágicas. Su papel en las trombosis en humanos no está claramente definido, si bien existe un trabajo en el que se ha relacionado los niveles plasmáticos elevados de FXI con el desarrollo de trombosis venosas71. Según este estudio, el 11% de todos los casos de trombosis venosas en la población general pueden ser atribuidos a valores elevados de FXI.
Indicaciones del estudio de hipercoagulabilidad
Existen una serie de indicaciones en las que este estudio se debe llevar a cabo de forma obligada (tabla 3). En estos casos una historia familiar de trombosis es un factor a tener en cuenta a la hora de realizar el estudio. Otras situaciones en las que parece estar indicado son las trombosis idiopáticas y las que aparecen en el curso del embarazo, el puerperio y el período neonatal, así como mujeres con pérdidas fetales de repetición, complicaciones gestacionales graves y aquellas que desarrollan trombosis tras el uso prolongado de anticoncepción oral72.

Este estudio se ha de posponer un mínimo de 2 meses desde la aparición de la trombosis para evitar el posible artefacto inducido por reactantes de fase aguda sobre diversos componentes hemostáticos. Además se ha de tener en cuenta el posible efecto del tratamiento recibido en el momento del estudio, ya que la heparina reduce la actividad funcional de la antitrombina y los anticoagulantes orales disminuyen la concentración de proteínas C y S73. Por tanto, y siempre que sea posible, se ha de realizar el estudio una vez completado el tratamiento anticoagulante y como mínimo 3 semanas después de haberlo suspendido.
La importancia de estos estudios se basa en que el conocimiento de una situación de hipercoagulabilidad puede orientar hacia la intensidad y duración del tratamiento anticoagulante (que puede ser indefinida) de forma más individualizada, marcar el rango de anticoagulación requerido, avisar del riesgo de complicaciones (como la necrosis cutánea por cumarínicos en el déficit de proteína C), sugerir la necesidad o intensidad de pautas de profilaxis y, en el caso de familiares asintomáticos de sujetos con hipercoagulabilidad, es importante identificar si el defecto está presente porque ello permitirá establecer pautas de profilaxis antitrombótica en situaciones de riesgo74.