




La hemocromatosis hereditaria (HH) se caracteriza por una absorción intestinal inadecuada del hierro contenido en la dieta que puede conducir, a través del depósito de este metal en diversos órganos, a la aparición de manifestaciones graves con una elevada morbimortalidad (fundamentalmente cirrosis, diabetes y miocardiopatía). Sin embargo, el inicio de un tratamiento adecuado antes de la aparición de estas complicaciones permite normalizar la expectativa de vida del paciente1. Por tanto, el objetivo primordial en el tratamiento de la HH debe ser alcanzar el diagnóstico de la manera más temprana posible. Ello exige un elevado índice de sospecha de esta entidad por parte del clínico, tarea no siempre fácil, dado que las manifestaciones tempranas de la enfermedad son, con frecuencia, leves e inespecíficas.
El descubrimiento reciente del gen HFE y de sus mutaciones ha modificado sustancialmente tanto el concepto como el tratamiento clínico de esta enfermedad2. En la actualidad, el análisis genético permite la confirmación del diagnóstico, evitando la biopsia hepática en muchos pacientes. Adicionalmente, simplifica el estudio familiar a partir de un caso índice y permite plantear la práctica de un cribado poblacional, ya sea en la población general o en grupos de elevado riesgo.
Genética y patogenia
En 1996 se descubrió un nuevo gen, denominado HFE2, situado en el brazo corto del cromosoma 6, próximo a la región HLA, que codifica una proteína HLA-I like. En este gen se detectaron dos mutaciones: la sustitución de cisteína por tirosina en el aminoácido 282 (C282Y) y la sustitución de histidina por aspartato en el aminoácido 63 (H63D). La mutación C282Y está presente en más del 85% de los cromosomas de pacientes diagnosticados de HH, mientras que sólo se detecta en el 3% de los cromosomas de individuos no portadores de la enfermedad2,3. Un porcentaje muy bajo de pacientes (entre un 0,5 y un 5%) son dobles heterocigotos o heterocigotos compuestos (es decir, heterocigotos para C282Y y para H63D)3. La asociación causal con otras posibles combinaciones de las mutaciones de HFE (heterocigotos para C282Y, heterocigotos para H63D y homocigotos para H63D) no está establecida y, en cualquier caso, su penetrancia sería muy baja3. Es interesante señalar que en algunos países del sur de Europa, sobre todo en Italia, se ha constatado una proporción sustancialmente inferior (alrededor del 60%) de homocigotos para C282Y4. En España, contrariamente a lo que podría suponerse dada nuestra proximidad con Italia, la situación es similar a la del norte de Europa, de forma que entre un 85 y un 90% de los pacientes con fenotipo HH son homocigotos para C282Y5-6. Otro aspecto a considerar es la prevalencia de la mutación C282Y en la población general, que se distribuye desigualmente en diferentes regiones, presentando una frecuencia elevada en poblaciones del centro y el norte de Europa (son homocigotos para C282Y una de cada 200-400 personas), mientras que es infrecuente o inexistente en otros grupos étnicos (orientales, africanos, melanesios, etc.)7. En el área mediterránea se han registrado las frecuencias alélicas más bajas dentro de población europea, especialmente en el sur de Italia y en Grecia. En España, de nuevo, los datos disponibles indican que aunque la prevalencia de la mutación C282Y en la población general es inferior a la del norte de Europa, prácticamente duplica a la obtenida en Italia o Grecia, de forma que uno de cada 1.100-1.400 individuos de la población general es homocigoto para C282Y5,8.
En la actualidad se conoce parcialmente el mecanismo patogénico de las mutaciones HFE en la HH. La mutación C282Y interrumpe la formación de un puente disulfuro en la molécula HFE, lo que impide la unión de ésta a ß2-microglobulina. Como consecuencia de ello, HFE no puede ser correctamente transportado desde el retículo endoplasmático a la superficie celular y, por tanto, no puede unirse al receptor de transferrina (TfR). La afinidad de TfR por la transferrina plasmática y la captación de hierro circulante plasmático por parte de la célula están moduladas precisamente por su unión a HFE. Así, en ausencia de la unión HFE-TfR, las células, fundamentalmente las de las criptas intestinales, no responden adecuadamente a los valores plasmáticos de hierro unido a transferrina. En último término, en el enterocito se produce la sobreexpresión de una proteína transportadora de metales divalentes (DMT1) que induce un aumento de la absorción intestinal de hierro9.
Es importante resaltar que entre un 10 y un 15% de pacientes con HH no presentan ninguna de las mutaciones del gen HFE. Por otro lado, se han detectado individuos homocigotos para C282Y que no presentan ninguna manifestación fenotípica de HH. Por ello, en la Reunión Internacional de Consenso celebrada en Sorrento en 199910 se acordó que el diagnóstico de HH sólo puede establecerse cuando existe alguna manifestación de la enfermedad, como mínimo una elevación del índice de saturación de transferrina (IST). Por tanto, la homocigosidad para C282Y en ausencia de alteración de los parámetros del hierro no es sinónimo de enfermedad, sino únicamente de predisposición o susceptibilidad genética.
Recientemente, se han descrito numerosas nuevas mutaciones del gen HFE que pueden explicar algunas de las HH no asociadas a homozigocidad para C282Y. Se han descrito, además, formas poco frecuentes de HH no relacionadas con las mutaciones HFE. Una de ellas, es la denominada hemocromatosis juvenil (HJ), en la que el depósito tisular de hierro tiene lugar antes de los 30 años de edad, afecta a ambos sexos por igual e induce la aparición de miocardiopatía e hipogonadismo graves. El defecto genético en la HJ se localiza en el cromosoma 1 y, aunque aún no se ha identificado el gen implicado, se le ha asignado el término HFE2. Otra forma de hemocromatosis es la asociada a diversas mutaciones del gen que codifica al segundo receptor de transferrina (TfR2), localizado en el cromosoma 7, y que se ha denominado HFE3. El gen TfR2 codifica una proteína transmembrana que se expresa predominantemente en el hígado, y cuya función es facilitar la captación celular del hierro unido a transferrina. Los sujetos homocigotos para alguna de estas mutaciones desarrollan una HH indistinguible de la que presentan pacientes con mutaciones del gen HFE11. Por último, también se ha descrito una forma familiar de sobrecarga férrica asociada a mutaciones del gen de la ferroportina, responsable de un cuadro de sobrecarga de hierro de predominio en las células reticuloendoteliales con un patrón de herencia autosómica dominante12. Los datos disponibles hasta el momento no permiten concluir que ninguna de estas nuevas formas de HH sea responsable de la enfermedad en pacientes que no presentan la mutación C282Y. Ello indica que tanto el gen HFE como otros genes implicados en el metabolismo férrico deben seguir siendo explorados en busca de nuevas mutaciones.
Clínica y situaciones de sospecha de HH
Como se ha observado, el término HH agrupa en realidad no a una sola entidad sino a una serie de trastornos genéticos caracterizados por el depósito tisular de hierro. La expresión completa de la enfermedad es multiorgánica, conduciendo a las manifestaciones clínicas clásicas de cirrosis hepática, diabetes, miocardiopatía, artropatía, hipogonadismo e hiperpigmentación cutánea (tabla 1). No obstante, durante los últimos años se ha ido constatando un cambio en la forma de presentación de la enfermedad, siendo cada vez más frecuente el diagnóstico precoz, incluso en pacientes asintomáticos13. Un aspecto crucial que cabe tener en cuenta es el de la penetrancia de la mutación C282Y. Se había considerado inicialmente que más del 50% de los homocigotos desarrollaban alguna manifestación clínica de la enfermedad. Sin embargo, estudios más recientes indican que la penetrancia real es probablemente muy inferior, estimándose que menos del 1% de los homocigotos desarrollarán una hemocromatosis franca14.

La sospecha de HH puede plantearse en diferentes escenarios clínicos (tabla 2) pero, a efectos prácticos, podemos agruparlos en dos grandes situaciones: a) búsqueda de casos en pacientes sintomáticos, y b) estudio familiar y cribado de población general, es decir, diagnóstico de individuos asintomáticos. En la búsqueda de casos, el diagnóstico será tardío si el signo o síntoma que condujo al mismo es consecuencia de alguna afección irreversible que acorte la expectativa de vida (p. ej., cirrosis, diabetes o miocardiopatía). Por ello, se ha de prestar especial atención a aquellas manifestaciones clínicas que, aun no siendo tempranas, permiten un diagnóstico precoz. Dos buenos ejemplos de estas últimas son la artropatía y la hipertransaminasemia. En muchas ocasiones, la sospecha se establece por el hallazgo casual de elevación de los parámetros del hierro en una analítica rutinaria o realizada por otro motivo. Obviamente, deberán buscarse otras posibles causas de sobrecarga de hierro diferentes de la HH (enfermedades hematológicas, politransfusión, etc.), pero sobre todo es importante descartar la presencia de otras hepatopatías (alcohólica, hepatitis vírica, porfiria cutánea tarda o esteatohepatitis no alcohólica). Estas entidades pueden cursar con alteración de los parámetros habituales del metabolismo del hierro e incluso pueden conducir a sobrecarga férrica hepática, que puede llegar a ser clínica, analítica e incluso histológicamente indistinguible de la HH.

Confirmación de la sospecha: pruebas bioquímicas (IST y ferritina)
Una vez considerada la posibilidad de HH, el primer paso para confirmar la sospecha consiste en demostrar la existencia de algún dato de alteración de los parámetros del metabolismo del hierro. Para ello, disponemos fundamentalmente de dos elementos: el índice de saturación de transferrina (IST) y la ferritina plasmática. El IST se obtiene del cociente entre la sideremia basal del paciente en ayunas y la capacidad total de fijación de hierro (TIBC en su acrónimo inglés). El IST se expresa como porcentaje y refleja hasta qué grado la transferrina del paciente se encuentra saturada de hierro en condiciones basales. La elevación del IST se produce muy pronto en el transcurso de la historia natural de la enfermedad, lo que la convierte en un dato casi universalmente presente en los pacientes con HH, con una cifra media alrededor del 70%, siendo superior al 90% en la mayor parte de los pacientes sintomáticos. Se considera que la sensibilidad y la especificidad del IST para el diagnóstico de HH son muy elevadas, superiores al 90%, con un valor predictivo positivo alrededor del 85%. No obstante, hay que señalar que estas cifras se han obtenido fundamentalmente a partir de estudios de población general, donde otras situaciones de sobrecarga férrica son relativamente infrecuentes. En otros contextos, como en la cirrosis hepática, por ejemplo, donde la propia hepatopatía cursa con frecuencia con una elevación del IST, su especificidad, y por consiguiente su valor predictivo positivo, es muy inferior. Las características del IST como prueba diagnóstica también dependen, lógicamente, del punto de corte considerado: cuanto menor sea éste mayor será la sensibilidad a expensas de la pérdida de especificidad, y viceversa. La tendencia de los últimos años, a medida que se ha perfilado el IST como mejor prueba diagnóstica inicial y ha aparecido el test genético (que aporta una gran especificidad como segunda prueba de confirmación), ha sido disminuir el umbral a fin de incrementar la sensibilidad. Un valor del 45% como punto de corte tiene una sensibilidad cercana al 100%15. Dada la variabilidad fisiológica del IST, especialmente notable en el período posprandial, a menudo se considera necesario demostrar una elevación del IST superior al 45%, al menos en dos determinaciones, asegurándonos de que se realiza la extracción sanguínea con el paciente en ayunas de 12 h.
A diferencia de lo que sucede con el IST, la elevación de la ferritina plasmática es un dato más tardío en la evolución de la HH y, por tanto, es una prueba menos sensible para su detección. Además, es menos específico, ya que se comporta de hecho como un reactante de fase aguda y puede elevarse en el contexto de numerosas enfermedades infecciosas, inflamatorias, neoplásicas y, muy característicamente, con el consumo de alcohol. Por tanto, la ferritina plasmática no es un buen método para la detección de HH y no tiene sentido determinarla de forma aislada, con este objetivo, sin acompañarse del IST. La mayor utilidad de determinar la ferritina, en el contexto del diagnóstico de la HH, estriba en que, a diferencia del IST que se satura precozmente, refleja de forma indirecta el grado de sobrecarga de hierro corporal y nos permite tomar algunas decisiones respecto al manejo del paciente, fundamentalmente la indicación de biopsia hepática y la necesidad de iniciar el tratamiento con flebotomías.
Confirmación del diagnóstico: biopsia hepática y estudio genético
Dado que, como se ha mencionado, los parámetros bioquímicos del metabolismo del hierro no son absolutamente precisos, se exige la realización de otra técnica que permita establecer de forma firme el diagnóstico. Tradicionalmente, el método considerado de referencia para el diagnóstico de HH ha sido la práctica de una biopsia hepática a fin de cuantificar el depósito de hierro en el parénquima hepático y calcular el índice de hierro hepático (IHH). La sobrecarga hepática de hierro puede demostrarse mediante la tinción de Perls, y adopta, sobre todo en las fases tempranas, un patrón característico consistente en una distribución predominantemente intrahepatocitaria con gradiente portocentral. No obstante, es preferible determinar la concentración de hierro en tejido hepático seco (µmol/g), que permite calcular el IHH (concentración de hierro en tejido hepático seco dividido por la edad del paciente en años). Otros métodos cualitativos o semicuantitativos para evaluar el grado de depósito de hierro hepático no son tan precisos. La utilidad del IHH se basa en la idea de que, a diferencia de lo que sucede en otras situaciones con sobrecarga hepática de hierro, el depósito férrico en la HH se produce de forma proporcional a la edad. Se ha considerado que un IHH >= 1,9 es prácticamente diagnóstico de HH16. Se sabe sin embargo, desde la aparición del test genético, que un número significativo de pacientes homocigotos para C282Y tienen un IHH inferior a 1,9; mientras que pacientes con hepatopatías avanzadas y negativos para C282Y pueden tener un IHH superior a 1,917.
En la actualidad, mediante análisis del ADN a partir de una muestra de sangre, puede determinarse con facilidad la presencia de las mutaciones C282Y y H63D del gen HFE. Desde un punto de vista práctico, dado que ambas mutaciones se hallan en desequilibrio de ligamiento en la práctica totalidad de los casos (son mutuamente excluyentes), la determinación de H63D puede reservarse sólo para los heterocigotos para C282Y. Como ya se ha comentado antes, se acepta que los pacientes homocigotos para C282Y con indicios de sobrecarga de hierro según los parámetros bioquímicos habituales pueden considerarse diagnosticados de HH e iniciar el tratamiento pertinente. En esta situación la biopsia hepática tiene únicamente un valor pronóstico a fin de determinar la presencia de cirrosis y/o fibrosis significativa. Durante los últimos años ha quedado claro que en pacientes menores de 40 años, sin indicios de afección hepática (con cifras de transaminasas normales y ausencia de hepatomegalia) y con concentraciones de ferritina sérica inferiores a 1.000 ng/ml puede excluirse la presencia de fibrosis significativa o cirrosis
y, por tanto, no es preciso practicar una biopsia hepática18. Por lo que respecta a los dobles heterocigotos (C282Y/H63D), aunque se reconoce que poseen un genotipo potencialmente responsable de la enfermedad, su penetrancia es muy baja y, por tanto, se aconseja la práctica siempre de biopsia hepática de confirmación. Por lo que respecta al resto de posibles combinaciones (heterocigotos para C282Y, heterocigotos u homocigotos para H63D y casos sin ninguna mutación de HFE), es preciso también la práctica de biopsia hepática para confirmar el diagnóstico.
Otros métodos de diagnóstico
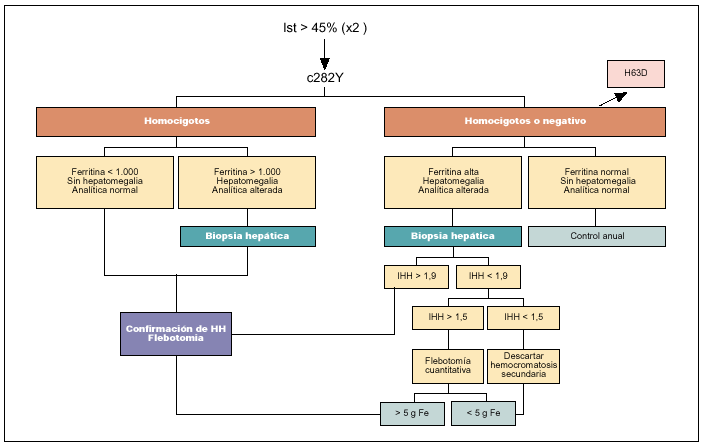
La determinación del número de flebotomías (por cada 500 ml de sangre extraída se eliminan 250 mg de hierro) necesarias para obtener la depleción de hierro (flebotomía cuantitativa) es otro método utilizado en el pasado para confirmar el diagnóstico, especialmente cuando por algún motivo no podía realizarse la biopsia hepática. Se basa en que los pacientes con HH precisan normalmente un mínimo de 20 flebotomías de 500 ml, es decir, la extracción de 5 g de hierro, para finalizar el tratamiento inicial. Actualmente, sólo tiene alguna utilidad en pacientes no homocigotos para C282Y y en los que no puede realizarse una biopsia hepática. Además, y a medida que el diagnóstico se realice más precozmente, cada vez serán más los pacientes con HH que precisen un número limitado de flebotomías para normalizar sus cifras de ferritina. Por lo que respecta a las técnicas de imagen, como la TC o la resonancia magnética, éstas carecen de la suficiente precisión para ser utilizadas como técnicas sistemáticas de diagnóstico que sustituyan a la biopsia hepática en el caso en que ésta sea necesaria. En la figura 1 se presenta el algoritmo diagnóstico aconsejado en la evaluación de un paciente con sospecha de HH.
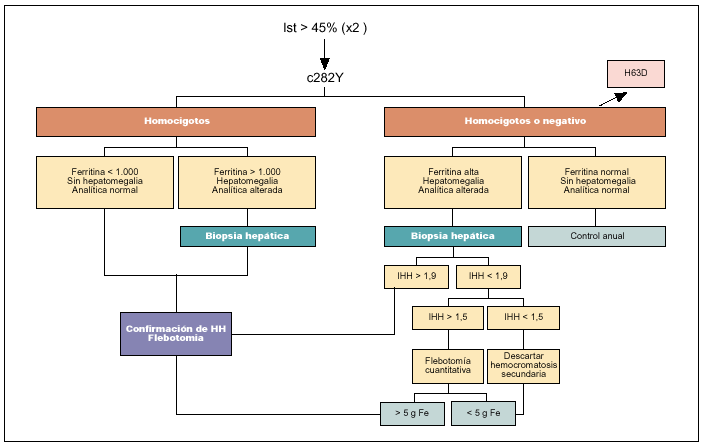
Fig. 1. Algoritmo diagnóstico de hemocromatosis en un caso individual.
Cribado familiar y poblacional
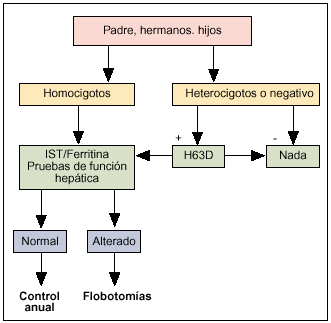
El estudio genético ha reemplazado al análisis del alelo HLA-A3, utilizado en la década de los ochenta, y simplifica considerablemente el estudio familiar en la mayor parte de los casos. En la figura 2 se incluye el algoritmo propuesto para el cribado de los familiares de primer grado de un caso índice. Partiendo de un caso índice homocigoto para C282Y es posible evaluar fácilmente el riesgo de sus familiares. Aquellos que sean, a su vez, homocigotos para C282Y deben ser considerados como potenciales portadores de la enfermedad. Por tanto, en ellos deberá evaluarse la posible expresión fenotípica mediante IST y ferritina, siguiendo el procedimiento diagnóstico descrito. Los familiares heterocigotos desarrollarán raramente una sintomatología clínica, aunque pueden transmitir la enfermedad (precisan, por tanto, un adecuado consejo genético), mientras que los no portadores pueden considerarse fuera de riesgo. Dado que en la HH asociada a HFE (a diferencia de la rara hemocromatosis juvenil) no se ha descrito la aparición de ningún tipo de síntoma antes de la edad adulta, se aconseja llevar a cabo el cribado en los niños solamente cuando alcancen la pubertad o la adolescencia. Una estrategia alternativa y más rápida para el estudio de los hijos de un caso índice es determinar el genotipo del otro padre/madre, ya que si en él está ausente la mutación C282Y, el máximo riesgo para los hijos sería ser heterocigotos.
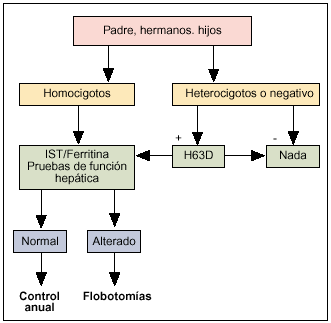
Fig. 2. Algoritmo de estudio familiar a partir de un homocigoto para C282Y.
La HH reúne las características adecuadas para realizar un cribado de la población general: a) es una enfermedad frecuente; b) se conoce la historia natural, existiendo una fase presintomática prolongada y detectable; c) existe un tratamiento eficaz y seguro que, instaurado en fase temprana, mejora claramente el pronóstico, y d) el método diagnóstico utilizado para el cribado puede ser aceptado por la mayoría de la población. Además, existen suficientes evidencias de que el cribado de la población general es coste-eficaz18. Aunque no está todavía estrictamente definida, se asume que el cribado de HH debe limitarse a poblaciones de origen étnico europeo (dada la rareza de la HH asociada a mutaciones de HFE en otros grupos) y realizarse a principios de la edad adulta, entre los 20 y los 30 años. Por lo que respecta a la estrategia a utilizar, el test genético ha sido evaluado de forma hipotética, y ha demostrado ser más útil aplicado como test de confirmación de los pacientes seleccionados en primera instancia por elevación de IST que utilizado como primer paso de la estrategia de cribado. A pesar de ello, el cribado de HH en la población general no se ha llevado a la práctica de forma generalizada y sistemática. Los motivos se derivan, fundamentalmente, de la indefinición todavía existente acerca de la definición de caso, de la penetrancia de los diferentes genotipos y de las posibles implicaciones éticas y sociales derivadas del diagnóstico de HH en personas asintomáticas.
Tratamiento
La extracción de sangre mediante flebotomías es el tratamiento de elección para la HH. Aunque ocasionalmente se han utilizado quelantes del hierro y eritrocitoaféresis, no hay ningún argumento de eficacia, seguridad o economía que justifique su uso. Como norma general, todos los individuos con genotipo compatible con HH y sobrecarga de hierro, así como aquellos con criterios clínico-histológicos de HH y sobrecarga de hierro con cualquier genotipo, son subsidiarios de recibir tratamiento, independientemente de que presenten o no sintomatología clínica, entendiendo por sobrecarga de hierro la presencia de ferritina plasmática por encima de los valores de normalidad. La edad o la presencia de una cirrosis descompensada no deben considerarse a priori contraindicaciones del tratamiento. Sólo en los raros casos de intolerancia a las flebotomías puede ensayarse tratamiento quelante. Habitualmente, se extraen entre 400 y 500 ml de sangre en cada flebotomía (ello supone la eliminación de 200-250 mg de hierro en cada extracción), con una periodicidad entre semanal y quincenal. Se mantiene esta pauta hasta que la ferritina plasmática desciende por debajo de 50 ng/ml19. Como monitorización del tratamiento debe utilizarse la determinación de hematocrito o hemoglobina antes de cada flebotomía, controlando los valores de ferritina únicamente cada 10 o 12 flebotomías. El IST no es un buen parámetro para el control del tratamiento, dado que se modifica de forma mucho más lenta que la ferritina. Posteriormente, deben realizarse flebotomías de mantenimiento cada 3 o 6 meses para mantener los valores de los parámetros del metabolismo del hierro dentro de los márgenes citados. En principio, este régimen debe mantenerse durante toda la vida, aunque en algunos pacientes, y por causa desconocida, la reacumulación de hierro tras la depleción inicial es muy leve e incluso inexistente. Como medidas adicionales, se aconseja evitar aquellos alimentos especialmente ricos en hierro (como el hígado y la carne roja), los suplementos orales de hierro y disminuir o suprimir el consumo de alcohol. No es necesario llevar a cabo una dieta estricta pobre en hierro. Debe evitarse, no obstante, el consumo de vitamina C. Por el contrario, beber té es beneficioso, ya que disminuye la absorción intestinal de hierro. Los antiinflamatorios no esteroideos son útiles en el tratamiento sintomático de la artropatía y se puede conseguir una mejoría de los síntomas de hipogonadismo con la administración de andrógenos.
El tratamiento con flebotomías, en general, es bien tolerado y su eficacia es absoluta en cuanto a conseguir la depleción de hierro. Asimismo, es capaz de prevenir la aparición de la mayor parte de las manifestaciones de la enfermedad, especialmente de las que llevan aparejadas una disminución de la expectativa de vida (cirrosis, diabetes y miocardiopatía), aunque en ocasiones algunos síntomas, como la astenia, las artralgias o la impotencia, pueden aparecer o empeorar después de iniciado. Por lo que respecta a los síntomas ya instaurados, el tratamiento con flebotomías es muy efectivo en la eliminación de la astenia y la hiperpigmentación cutánea, así como para normalizar la hipertransaminasemia, mientras que el efecto es menor sobre la artropatía, la diabetes, la miocardiopatía y la impotencia. La cirrosis, por su condición de lesión irreversible, no es susceptible de desaparecer con las flebotomías, aunque es interesante destacar que dicho tratamiento tiene un efecto beneficioso sobre la hipertensión portal, que se traduce en una disminución del riesgo de hemorragia por varices esofágicas. El tratamiento adecuado no previene, sin embargo, el riesgo de desarrollar un hepatocarcinoma una vez instaurada la cirrosis.