







La fiebre mediterránea familiar (FMF) es una enfermedad hereditaria transmitida de forma autosómica recesiva, cuyas características clínicas más sobresalientes son los episodios agudos de fiebre y serositis (peritoneo, pleura, articulaciones y túnica vaginal del testículo), que duran desde horas hasta 2 o 3 días y ceden espontáneamente, permaneciendo el paciente asintomático entre las crisis. Afecta especialmente a los descendientes de judíos sefardíes, askenazíes, armenios, árabes y turcos, y la amiloidosis es la principal causa de muerte.
Genética
Los estudios realizados en un importante número de pacientes con FMF y de sus familiares permitió concluir que esta enfermedad se transmite con carácter autosómico recesivo. Si ambos progenitores son portadores de un rasgo autosómico recesivo, existe un 25% de probabilidades de que un hijo herede los dos genes anormales de sus padres y desarrolle la enfermedad. En ocasiones, la FMF aparece en determinadas familias en dos o más generaciones, lo que sugiere una transmisión autosómica dominante, posiblemente explicada por la elevada tasa de consanguinidad en las mismas, al casarse los pacientes con portadores asintomáticos. La tasa de portadores entre la población judía no es uniforme y depende de sus orígenes.
En 1992 se localizó el gen de la FMF, denominado MEFV (abreviatura de mediterranean fever), en el brazo corto del cromosoma 16. En 1997 dos grupos independientes consiguen clonar y caracterizar el MEFV, denominándose a la proteína resultante de la transcripción de dicho gen marenostrina (del latín, Mare Nostrum) por parte del Consorcio francés-israelí, y pirina (por su relación con la pirexia-fiebre) por parte del Consorcio Internacional. Dicho gen se expresa especialmente en las células mieloides, y aumentan su expresión durante la diferenciación celular, sobre todo en el neutrófilo maduro y en el monocito actuando como estimuladores el interferón gamma y el factor de necrosis tumoral. No se conoce la función específica de esta proteína, pero parece que regula los procesos inflamatorios relacionados con los neutrófilos.
Los primeros estudios demostraron que más del 80% de los pacientes con FMF presentaban mutaciones localizadas en un tramo de 46 aminoácidos codificados en el exón 10 del gen MEFV. Inicialmente, se describieron cuatro mutaciones en dicho exón y actualmente ha sido identificado un total de 28 mutaciones. Sus expresiones fenotípicas son variadas, y se han identificado algunas cuyos síntomas son tan mínimos o infrecuentes que podrían confundirse con variaciones de la normalidad.
Epidemiología
La FMF se manifiesta especialmente en algunos grupos étnicos originarios del litoral mediterráneo, como judíos sefardíes, armenios, turcos, árabes y, menos comúnmente, griegos e italianos. En el norte de Europa y en Estados Unidos está descrita en judíos askenazíes. En nuestro país se ha identificado una agrupación de enfermos, entre los descendientes de los judíos conversos en Mallorca, objetivándose en el 30% un haplotipo genético similar al de pacientes de otras zonas geográficas. De manera aislada, se ha diagnosticado a pacientes de Girona, Toledo, Madrid, Andalucía y otras zonas españolas.
Se describen dos fenotipos con características clínicas y evolutivas diferentes. En el fenotipo I, el más frecuente, las manifestaciones clínicas preceden a la amiloidosis cuando ésta se presenta. Por lo general, se observa una disminución en el número y la intensidad de las crisis conforme aumenta la edad del paciente. En el fenotipo II, la amiloidosis es la primera o única manifestación de la enfermedad. Se ha descrito una correlación entre el genotipo y el fenotipo. En los individuos homozigotos para la mutación M694V, la FMF se caracteriza por un inicio más temprano con episodios más frecuentes y con una mayor asociación con la amiloidosis. El pronóstico de la enfermedad está íntimamente relacionado con la aparición de amiloidosis. Aparte de las mutaciones del gen MEFV, se conoce que los polimorfismos en el gen del amiloide sérico A aumenta dicha susceptibilidad.
Etiopatogenia
La patogenia de esta enfermedad sigue siendo confusa y existen varias teorías que se han elaborado para explicar su diversa sintomatología. Hay suficiente evidencia como para considerar al neutrófilo como el efector de la respuesta inflamatoria en las serosas; de hecho, en diferentes tratados se incluye dentro de los trastornos hereditarios de la función del neutrófilo. Apoyan esta hipótesis el predominio de neutrófilos en las serosas durante los ataques agudos, la efectividad de la colchicina en el tratamiento y prevención de las crisis por sus efectos supresores de la fagocitosis y la quimiotaxis, así como la expresión casi única de la marenostrina/pirina en los neutrófilos circulantes. Se cree que dicha proteína actúa como un autorregulador supresor de la respuesta de estas células frente a estímulos inflamatorios, interfiriendo con la función de su citosqueleto. Su defecto podría originar una activación y migración incontrolada de los neutrófilos.
También se ha postulado el déficit de un inhibidor de una de las fracciones de la activación del complemento, el C5a. La base de esta hipótesis se sustenta en la existencia, durante la crisis, de un déficit en la actividad de la enzima inactivadora del factor quimiotáctico del complemento C5a. Diferentes estudios han demostrado una disminución de la actividad de esta enzima en dichos pacientes, tanto en el líquido sinovial como en el peritoneo, lo que permitiría la persistencia del factor quimiotáctico y la llegada de los neutrófilos con liberación de sus productos, que culminan en la reacción inflamatoria que daría lugar a los episodios esporádicos de fiebre y serositis. Inicialmente, se consideró que la marenostrina/pirina era la enzima inactivadora, pero ya se conoce que dicho inhibidor del complemento tiene una estructura diferente de la marenostrina/pirina.
Anatomía patológica
El hallazgo fundamental durante las crisis de la FMF es la reacción inflamatoria de las serosas peritoneo, pleura, pericardio, sinovia y túnica vaginal del testículo con una infiltración masiva de polimorfonucleares en los tejidos afectados, secundaria a un aumento de la
actividad quimiotáctica de los neutrófilos y a su activación.
La amiloidosis que desarrollan estos pacientes es de tipo reactivo, con la proteína AA en las fibras amiloides, y su génesis se relaciona con los valores elevados de la proteína sérica SAA (serum amyloide A), que actúa como reactante agudo durante los ataques de fiebre y que se normaliza entre ellos. Sin embargo, a pesar de la frecuencia de la amiloidosis sistémica en la FMF, la relación entre ésta y la amiloidosis no está clara. El desarrollo de la amiloidosis no se correlaciona bien con el número o intensidad de los ataques febriles, e incluso algunos miembros de las familias con FMF desarrollan una amiloidosis sin episodios febriles, lo que se ha designado fenotipo II. Además, la frecuencia de esta complicación no es igual entre los diferentes grupos de riesgo, lo que sugiere para algunos autores que son dos rasgos diferentes con base genética distinta. La amiloidosis se visualiza preferentemente en los glomérulos renales. Los depósitos vasculares que infiltran la íntima y la media de las diferentes arteriolas son frecuentes y universales, pero muy raramente producen una disfunción orgánica. También hay depósitos de amiloide en el bazo, las suprarrenales, la glándula tiroides y, con menor frecuencia, en otros órganos. Sorprende la ausencia de afectación del hígado, ya que los depósitos de amiloide están limitados a las estructuras vasculares de los tractos de la porta.
Cuadro clínico
Los síntomas se inician en la primera década de la vida en el 50% de los casos, antes de los 20 años en el 80%, y después de los 30 años en el 5%. La duración y la frecuencia de los ataques varían extraordinariamente incluso en el mismo paciente. Por lo común, la crisis dura de horas a días, repitiéndose de una manera periódica. La sintomatología se evidencia sólo durante las crisis. Las características generales se resumen en
las tablas 1 y 2.


Fiebre
La fiebre está presente en casi todos los casos y, en la mayoría de ellos, la temperatura llega a los 38-40 °C, aunque los ataques leves pueden cursar con febrícula; por lo general, va precedida de escalofríos y dolor abdominal, articular o torácico. De forma excepcional, se prolonga más de una semana y suele disminuir bruscamente.
El dolor abdominal está presente en el 95% de los casos; se localiza especialmente en el hipocondrio y la fosa ilíaca derecha y después se extiende de manera difusa por todo el abdomen. Varía desde una ligera molestia hasta un dolor muy intenso, que recuerda las manifestaciones de una perforación de víscera hueca. Disminuye de forma gradual y desaparece después de la fiebre. Los vómitos y el estreñimiento son frecuentes.
Dolor torácico
El dolor torácico puede estar causado por un derrame pleural o pericárdico; el primero se presenta en un momento u otro de las crisis, aproximadamente en la mitad de los casos, su comienzo es brusco y desaparece de forma paulatina en un intervalo de horas; en casos excepcionales se prolonga durante varios días. El dolor pericárdico es menos frecuente.
Artritis
Las manifestaciones articulares varían en su incidencia según el grupo étnico y se presentan en forma aguda o crónica. La forma aguda se manifiesta como monoartritis u oligoartritis asimétrica de comienzo brusco, que cede lentamente, aunque puede durar varios días. La forma crónica tiene una incidencia más baja, dura semanas o meses; puede aparecer independientemente del resto de las manifestaciones de la FMF y, a menudo, la recuperación es completa. Ocasionalmente, puede presentarse como una poliartritis migratoria.
Afección cutánea
Las manifestaciones cutáneas se pueden presentar en forma de eritema erisipeloide localizado en las extremidades inferiores o como lesiones nodulares.
Otras manifestaciones
Con menor frecuencia se observan orquitis autolimitadas, meningitis asépticas recurrentes y mialgias y, en ocasiones, puede palparse una esplenomegalia.
Se ha descrito una asociación entre la FMF y ciertas vasculitis, como son la púrpura de Schönlein-Henoch (PSH) y la poliarteritis nudosa. La frecuencia de la PSH entre 207 pacientes turcos con FMF fue del 7,2%, mientras que en la población pediátrica general visitada durante el mismo período de 10 años fue del 0,8%. Algo similar en cuanto a la frecuencia ocurre con la poliarteritis nudosa con la característica añadida de que afecta a gente joven.
Durante las crisis existe leucocitosis, una elevación de la VSG y un aumento de la proteína C reactiva y del fibrinógeno sérico, que se normalizan en las intercrisis. Los exámenes radiológico y ecográfico pueden demostrar la existencia de derrame pleural o pericárdico en los casos en que se presentan.
La frecuencia de la amiloidosis difiere entre los diversos grupos étnicos y depende de si los pacientes han sido tratados con colchicina. Suele depositarse en los riñones, originándose un síndrome nefrótico y una insuficiencia renal; afecta más raramente al tracto gastrointestinal y otras zonas. En la mayoría de los casos se desarrolla antes de los 40 años de edad.
Diagnóstico
El diagnóstico de la FMF se basa fundamentalmente en la historia clínica del paciente, el patrón evolutivo de sus síntomas y signos, sus características étnicas, la historia familiar y la respuesta a la colchicina.
Los episodios recurrentes con períodos asintomáticos y de normalidad entre ellos son una de sus características más sobresalientes y la razón de que se incluyan en lo que Reiman denominó, en 1940, «enfermedades periódicas». En la actualidad, en que se considera que los patrones febriles tienen poco significado clínico, dicho concepto no tiene más valor que plantear a los clínicos diferentes diagnósticos diferenciales de procesos de fiebre prolongada que evolucionan de forma «recurrente» o «episódica», independientemente del tiempo (meses o años) en que ocurra dicha recurrencia.
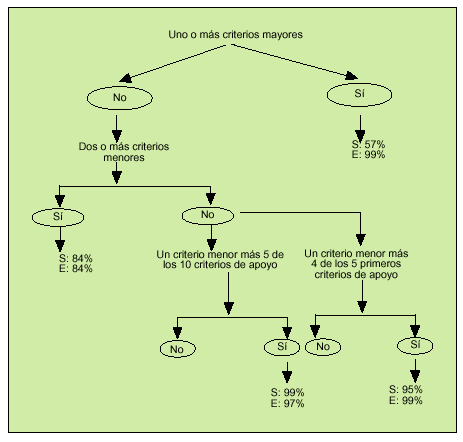
En 1997, Livneh et al publicaron unos criterios para el diagnóstico de la FMF (tabla 3) que tienen una sensibilidad del 95% y una especificidad del 99% para el diagnóstico, a partir de los cuales construyeron un algoritmo (fig. 1). En ocasiones y ante situaciones con sospecha fundamentada, muchos autores optan directamente por el tratamiento con colchicina y la valoración de la respuesta.

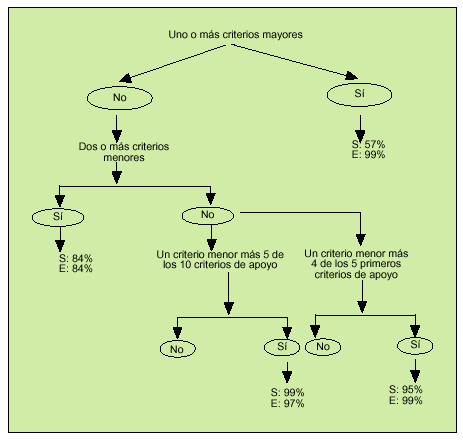
Fig. 1. Algoritmo diagnóstico de fiebre mediterránea familiar. S: sensibilidad; E: especificidad. (Adaptado de Livneh, et al. Arthritis Rheum 1997; 40:1879.)
La clonación del gen de la FMF ha permitido disponer de una prueba diagnóstica nueva y fiable para esta enfermedad. Las mutaciones ocurren en ambos alelos en el 70% de los casos típicos, mientras que en el 30% restante puede detectarse una sola mutación o ninguna. El clínico debe saber que los laboratorios de genética sólo detectan las mutaciones más frecuentes, mientras que el resto puede pasar inadvertido. Incluso existe evidencia de la no penetrancia en algunas mutaciones, es decir, que pueden existir dos mutantes en ausencia de enfermedad. De todas formas, el diagnóstico genético puede ser utilizado en las formas dudosas o menos típicas en las que el índice de sospecha es alto, o en determinadas formas clínicas, especialmente la amiloidosis renal que aparece en ausencia de ataques agudos y al inicio del proceso, por las implicaciones familiares que dicho diagnóstico lleva. De cualquier manera, quedan por definir las indicaciones y la utilidad del estudio genético. En la tabla 4 se indica la contribución actual del estudio genético en el diagnóstico de la enfermedad.

La dificultad en el diagnóstico del proceso depende de su edad de comienzo, el tiempo de enfermedad transcurrido, su historia familar y el grado de sospecha del médico. En la edad pediátrica requiere un amplio diagnóstico diferencial, especialmente con procesos infecciosos intercurrentes o localizados, tumores y enfermedades autoinmunes; además, se ha descrito en los niños un síndrome de fiebre periódica (periodic fever aphthous stomatitis, pharingitis and adenopathy) que cursa con estomatitis aftosa, faringitis y adenitis cervical dolorosa, que duran unos 4 días y recurren cada 2-8 semanas. En general, desaparece sin secuelas entre los 4 y los 8 años, no existe historia familiar y se desconoce si es un defecto genético. Especial mención merece la afección en la edad pediátrica de la túnica vaginal del testículo, cuyos espisodios inflamatorios se deben diferenciar de la torsión del testículo, de las orquiepidedimitis infecciosas e incluso de las vasculitis tipo PAN.
Un problema diagnóstico importante son los cuadros abdominales agudos que requieren un amplio diagnóstico diferencial, tanto de las enfermedades quirúrgicas abdominales como de las no quirúrgicas. Entre las fundamentales se encuentran las apendicitis agudas, la colecistitis aguda, la perforación abdominal, el brote ulceroso agudo y las pancreatitis recurrentes, y entre los «abdómenes agudos médicos», la diabetes mellitus descompensada, la insuficiencia suprarrenal aguda, las crisis hemolíticas, la porfiria aguda intermitente, la intoxicación por plomo, el edema angioneurótico familiar, el LES con vasculitis, la hipertrigliceridemia, la migraña abdominal y otras situaciones menos frecuentes, entre las que se incluye el resto de las fiebres periódicas. La indicación de laparotomía es bastante frecuente si no se establece el diagnóstico de la enfermedad correctamente en la primera crisis, e incluso conociendo el diagnóstico, no siempre es fácil diferenciarla de cuadros quirúrgicos como las apendicitis, por lo que algunos autores preconizan la apendicectomía laparoscópica profiláctica, una vez establecido el diagnóstico de FMF.
A medida que transcurre el tiempo, las causas infecciosas o neoplásicas van perdiendo peso. Con las nuevas técnicas de imagen sólo un limitado número de procesos infecciosos o tumorales son capaces de producir unos patrones febriles similares, aunque no se debe olvidar procesos como los mixomas auriculares. En general, el diagnóstico diferencial se establece con la artritis reumatoide juvenil, la enfermedad de Still del adulto, la enfermedad de Crohn, el síndrome de Behçet, la policondritis recidivante, la fiebre secundaria a la toma intermitente de drogas, la neutropenia cíclica, algunas enfermedades granulomatosas y la fiebre ficticia.
Junto a la FMF, los otros dos síndromes más conocidos, y cuyos defectos genéticos se conocen, son el TRAPS (tumor necrosis factor receptor-associated periodic syndrome) y el HIDS (hiperinmunoglobulininemia D). El TRAPS es una enfermedad hereditaria dominante poco frecuente donde se incluyen diferentes entidades antiguas, conocidas como fiebre hiberniana familiar, fiebre periódica familiar benigna autosómica dominante y fiebre periódica autosómica dominante con amiloidosis. La HIDS es una fiebre periódica heredada recesivamente y cuya característica sine qua non es la elevación persistente de la IgD acompañada en el 80% de aumento de la IgA. Se han descrito más de 100 casos en el norte de Europa y su alteración molecular es una mutación en el gen MVK (mevalonato kinasa) que codifica la mevalonato cinasa que interviene en la síntesis del colesterol. Las características diferenciales entre las fiebres periódicas hereditarias se incluyen en la tabla 5.

Tratamiento
El fármaco a utilizar en esta enfermedad es la colchicina que, a dosis de 0,6-1,8 mg/día, es capaz de prevenir las crisis agudas y la aparición de amiloidosis, estabilizando la filtración glomerular en pacientes con proteinuria moderada. Si existe un síndrome nefrótico, se puede prevenir el progreso de la enfermedad y una reducción de la excreción de proteínas, para lo que se requiere unas dosis de colchicina algo mayores de 1,5-2,0 mg/día y comenzar la terapéutica antes de que la concentración de creatinina en plasma alcance los 1,5 mg/dl (132 mmol/l); el beneficio es gradual y se requiere un período entre uno y dos años.
En los pacientes con insuficiencia renal avanzada, la hemodiálisis permite mantener con vida a estos enfermos, e incluso durante el tratamiento con hemodiálisis, la administración de colchicina disminuye la frecuencia de las crisis paroxísticas. La amiloidosis no constituye una contraindicación para los trasplantes renales, pero se debe continuar el tratamiento con colchicina para prevenir o retrasar la aparición de la amiloidosis en el riñón trasplantado y en otros órganos.
Dado que la FMF se puede presentar en la infancia, está indicado comenzar el tratamiento con colchicina, habiéndose demostrado que su uso prolongado no afecta ni al peso ni a las curvas de crecimiento. Se puede continuar el uso de la colchicina durante el embarazo, advirtiendo a estas mujeres de la necesidad de realizar una amniocentesis para el estudio del cariotipo. En los pacientes que no se benefician de la colchicina se han ensayado tratamientos con interferón alfa, abortando los ataques con una dosis única de 3 a 10 millones de U s.c. Se ha descrito un único caso de respuesta al bloqueador alfa prazosín.
Los efectos secundarios de la colchicina a la dosis de 1-2 mg/día se reducen al tracto digestivo en forma de náuseas o diarreas, que se resuelven al disminuir la dosis. En la médula ósea pueden aparecer manifestaciones transitorias de pancitopenia, y es extremadamente raro a las dosis terapéuticas objetivar neuropatía o miositis. Se ha descrito azoospermia y oligospermia reversibles.
Bibliografía general
Babior BM, Matzner Y. The familial Mediterranean fever gene-cloned at last. N Engl J Med 1997;337:1548-9.
Ben-Chetrit E, Levy M. Colchicine prophylaxis in familial Mediterranean fever: reappraisal after 15 years. Semin Atrhritis Rheum 1991;20:241.
Ben-Chetrit E, Levy M. Familial Mediterranean Fever. Lancet 1998; 351:659-64.
Booth D R, Gillmore J D, Lachmann H J, Booth S E, Bybee A, Soytürk M, et al. The genetic basis of autosomal dominant familial Mediterranean fever Q. J Med 2000;93:217-21.
Buades Reines J, Aguirre Errasti C. Fiebre mediterránea familiar. Diagnóstico y tratamiento. Med Clin (Barc) 2001;117:142-6.
Drenth JPH, Van der Meer JWM. Hereditary periodic fever. N Engl J Med 2001;345:1748-57.
Kataoka H, Jumagai H, Hani H. Treating familial Mediterranean fever with prazosin hydrochloride [letter]. Ann Intern Med 1998; 129:424.
Knockaert DC, Vanneste LJ, Bobbaers HJ. Recurrent or episodic fever of unkown origin. Review of 45 cases and survey of the literature. Medicine (Baltimiro) 1993;72:184-96.
Livneh A, Drenth JPH, Grateau G. Familial Mediterranean fever and hyperimmunoglobulinemia D syndrome: two diseases with distinct clinical, serologic and genetic features. J Rheumatol 1997;24:1558.
Liveneh A, Langevitz P. Diagnostic and treatment concerns in familial Mediterranean fever. Best Pract Res Clin Rheumatol 2000;14: 477-98.
Livneh A, Langevitz P, Zemer D, Zaks N, Kees S, Lidar T, et al. Criteria for the diagnosis of familial Mediterranean fever. Arthritis Rheum 1997;40:1879.
Matzner Y, Abedat S, Shapiro E, Shlomit E, Bar-Gil-Shitrit A, Stepensky P. Expresion of the familial Mediterranean fever gene and activity of the C5a inhibitor in human primary fibroblast cultures. Blood 2000;96:727-31.
McDermott MF, Aksentijevich I, Galon J, McDermott EM, Ogunkolade BW, Centola M, et al. Germline mutations in the extracellular domains of the 55kDa TNF receptor, TNRF1, define a family of dominantly inherited autoinflammatory syndromes. Cell 1999;97: 133-44.
McDermott EM, Smillie DM, Powell RJ. Clinical spectrum of familial Hibernian fever: A 14-year follow-up study of the index case and extended family. Mayo Cliln Proc 1997;72:806-17.
Mimouni A, Magal N, Stoffman N, Shohat T, Minasian A, Krasnov M, et al. Familial Mediterranean fever: effects of genotype and ethnicity on inflammatory attacks and amyloidosis. Pediatrics 2000;105:1-7.
Özdogan H, Arisoy N, Kasapcapur Ö. Vasculitis in Familial Mediterranean Fever. J Rheumathol 1997;24:323-7.
Padeh S, Brezniak N, Zemer D. Periodic fever, aphthous stomatitis, pharingitis and adenophathy syndrome: clinical characteristics and outcome. J Pediatr 1999;135:98-101.
The French FMF Consortium. A candidate gene for familial Mediterranean fever. Nat Genet 1997;17:25.
The International FMF Consortium. Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. Cell 1997;90:797.
Tunca M, Tankiurt E, Akbaylar Akpinar H, Akar S, Hizli N, Gonen O. The efficacy of interferon alpha on colchicine-resistant familial Mediterranean fever attacks: a pilot study. Br J Rheumatol 1997; 36:1005.
Zemer D, Pras M, Sohar E, Modan M, Cabili S, Gafni J. Colchicine in the prevention and treatment of the amyloidosis of familial mediterranean fever. N Engl J Med 1986;314:1001-5.