



La esclerosis lateral amiotrófica (ELA) es un trastorno neurodegenerativo del grupo de enfermedades del sistema motor, cuya etiología se desconoce y sin terapéutica eficaz. La sobrevida es dos a cinco años después del inicio. En este reporte se documentan las características fisiopatológicas y clínicas de la ELA, así como las opciones terapéuticas actualmente disponibles. Se realizó búsqueda electrónica exhaustiva y revisión de la literatura concerniente a la ELA, con énfasis particular en estudios que incluyeron pacientes tratados con terapia celular, por cualquier vía de administración, y en los resultados obtenidos con esta forma de tratamiento. Se analizó la respuesta con Riluzole y Litio, al igual que medicamentos en fase III de investigación. El tratamiento farmacológico ofrece beneficios mínimos, el Riluzole incrementa la sobrevida en meses. El dextropramipexol (actualmente en fase III) parece ser prometedor. Estudios en animales y humanos han demostrado que la terapia celular es factible y segura, en algunos casos conduciendo a mejoría significativa de la función motora. Sin embargo, no están definidas con precisión la vía óptima de administración, sea esta intraparenquimatosa, intraespinal, intravenosa y/o intratecal, ni el número de procedimientos necesarios para obtener una respuesta positiva. El tratamiento farmacológico de los pacientes con ELA ofrece pobres resultados. La terapia celular es una estrategia terapéutica prometedora en ELA, donde podría modificar la evolución y el pronóstico de este trastorno. Actualmente están en desarrollo protocolos para evaluar esta terapia.
Amyotrophic Lateral Sclerosis (ALS) is a neurodegenerative disorder that belongs to the motoneuron diseases. Its etiology is unknown with no definitive treatment available. Survival is two to five years after diagnosis. In this report, we describe its physiopathology and clinical features, as well as the current therapeutic options. We conducted a search on electronic databases and revised pertinent ALS literature, with particular attention to the studies describing therapeutic modalities such as Riluzole, Lithium and stem cell therapy, as well as the results obtained with these treatment alternatives. After reviewing pharmacological therapeutic options, at the present time, medications used in ALS offer poor results, with no foreseeable new drugs in the near future. Dextropramipexole is a phase III promising drug. Studies in animal models and in humans show that treatment with autologous stem cells is feasible and effective, in some cases leading to significant improvement in motor function. However, the optimal administration route, be it intracerebral, intraspinal, intravenous, and / or intrathecal, and the number of procedures needed to obtain a significant improvement in motor symptoms have not been defined. Pharmacologic agents for treatment of ALS offer unsatisfactory results. Autologous stem cell therapy appears as a potential therapeutic strategy in ALS that may modify outcome and prognosis. Several cell therapy protocols are currently in progress in different countries.
Introducción
La Esclerosis Lateral Amiotrófica (ELA), también conocida como enfermedad de Lou Gehrig (EUA), enfermedad de Charcot (Francia), enfermedad de Stronza (Italia) o de Neurona Motora (Inglaterra), es un trastorno neurodegenerativo clasificado dentro del grupo de las enfermedades de neurona motora. Consiste en la pérdida progresiva de motoneuronas, localizadas en la corteza cerebral y en el asta anterior de la médula espinal, cuyo resultado es la alteración de la función de los músculos voluntarios.1 Esta patología fue descrita por primera vez, en 1869 por Jean Marie Charcot y Alexis Joffroy en la Salpetriére, Charcot la denominó "Síndrome fatal".2
La ELA es un término descriptivo, esclerosis «endurecimiento», refiriéndose a la cicatriz que se forma en el tejido nervioso cuando éste se degenera, lateral de acuerdo a la topografía de afección en el cordón lateral de la medula espinal, afectando el tracto corticoespinal que transmite información de la neurona motora superior (NMS) hacia el asta anterior, y amiotrófica para describir la atrofia muscular que se produce por la muerte de la neurona motora inferior (NMI).
La ELA es una enfermedad progresiva, incapacitante y finalmente mortal, de etiología desconocida. Es más frecuente en el género masculino, a razón de 1.5:1 con respecto a las mujeres. Sin embargo, en mayores de 70 años se presenta con igual frecuencia en ambos géneros.2,3
La incidencia varía de uno a dos por cada 100 000 habitantes y aumenta con la edad,4 mientras que la prevalencia es de cuatro a seis casos por 100,000 habitantes. Se estima que existen alrededor de 120 000 personas afectadas por ELA en el mundo, lo que se traduce en 328 personas diagnosticadas y documentadas por día, de las cuales 30 000 habitan en Estados Unidos de América, donde se diagnostican alrededor de 5 000 nuevos pacientes por año.1 En España, alrededor de 4 000 personas padecen esta enfermedad, y cada año son diagnosticados aproximadamente 900 casos nuevos de ELA,3 lo que equivale a entre dos y tres pacientes diagnosticados por día.
Atletas famosos han padecido ELA, y diversos estudios epidemiológicos han señalado la actividad física de alto rendimiento como un factor de riesgo para desarrollar ELA en personas delgadas y atletas. Entre los atletas profesionales afligidos por esta enfermedad se cuentan Lou Gehrig (jugador profesional de beisbol) Ezzard Charles (campeón de peso pesado en box), Catfish Hunter (jugador de beisbol), Jacob Javits (jugador de tenis), David Niven (marinero de competencia), Stefano Borgonovo (futbolista del Milán), entre otros.5 Actualmente, se presume la existencia de 43 casos de ELA en jugadores profesionales de fútbol, lo cual en Inglaterra se ha relacionado con la presencia de cianobacterias en el pasto de la cancha, sin evidencia disponible de ello.
La edad de presentación se encuentra entre los 40 y 60 años, con edad promedio al diagnóstico de 54.4 años para las mujeres y 53.1 años para los hombres.6 Se ha reportado un aumento en la sobrevida cuando esta enfermedad ocurre en personas jóvenes, destacando como ejemplo el caso de Stephen Hawking, el físico teórico contemporáneo más importante, ya que fue diagnosticado a los 21 años y actualmente tiene 48 años con diagnóstico de ELA.1
Fisiopatología
La enfermedad se ha dividido en dos grandes vertientes: la mutacional, implicada en el 5% a 10% de los casos, y la multifactorial en el 90% a 95%. Entre las primeras, se encuentran alrededor de 100 mutaciones en el gen de la enzima Superóxido Dismutasa Cu-Zn Dependiente (SOD1), descrito por primera vez en 1993. SOD1 se codifica en el cromosoma 21q22 y su carácter hereditario es autosómico dominante, encontrándose únicamente en el 20% de los casos, el resto se vincula con otros cromosomas como 2q33-35, 15q15, 15q21, 9q34, Xp11 y Xp21, los primeros cuatro con carácter hereditario autosómico recesivo, mientras que los últimos dos están ligados al sexo.4
Para explicar las formas esporádicas de ELA existen actualmente cinco hipótesis sobre su origen, las cuales son: 1) excitotóxica, 2) stress oxidativo, 3) inmunológica, 4) trófica e 5) infecciosa.4
El mecanismo común implicado en todas las hipótesis mencionadas es el exceso de glutamato, un neurotransmisor excitador que abunda en el sistema nervioso central (SNC) y que resulta tóxico para las neuronas cuando se acumula, debido a que ocasiona entrada excesiva de Ca+2 a través de receptores AMPA (ácido propiónico de alfa amino 3-hidroxi 5-metil 4-isoxazol). En los pacientes con ELA estos receptores carecen de la subunidad R3 (fenilo o un anillo heteroarilo de cinco o seis eslabones) y receptores NMDA (N-metil-D-aspartato). La constante activación de estos receptores despolariza la membrana de las motoneuronas, haciendo que los canales de Ca+2 se mantengan abiertos, causando un mayor ingreso de éste.4 El Ca+2 libre en el citoplasma de la neurona genera radicales libres mediante la activación de la enzima óxido nítrico (ON) sintetasa, activando proteasas, fosfolipasas y endonucleasas, resultando en una desnaturalización del ADN y muerte celular.
La acumulación del glutamato se ha relacionado con una falla en los transportadores EAAT2 (Excitatory Amino Acid Transporter 2, por sus siglas en Inglés).4
Los mecanismos excitotóxico y de estrés oxidativo están ligados a la misma hipótesis, la cual señala que el daño es debido a la acumulación de moléculas prooxidativas OH (hidróxido) y O2 (oxígeno) principalmente, que generan peroxidación de los lípidos y proteínas de las membranas y organelos intracitoplasmáticos de las neuronas.4
La causa trófica postula una falla en la acción o producción de uno o más factores tróficos ocasionando muerte neuronal. Como causa infecciosa se han señalado las infecciones por adenovirus, mientras otras teorías indican que los mecanismos de origen inmunológico se deben a la existencia de anticuerpos dirigidos contra los canales de Ca+2, ocasionando alteraciones en la transmisión neuromuscular.4
Etiología
Se desconoce el evento inicial que desencadena los complejos cambios neuronales que conducen al desarrollo de la ELA. Las siguientes son algunas de las posibles causas propuestas para explicar el desarrollo de esta enfermedad:4
- Alteraciones en el metabolismo del glutamato, un aminoácido esencial en la conducción del impulso nervioso.
- Sensibilidad aumentada al daño neuronal por productos tóxicos del metabolismo (daño hidroxiradical).
- Reducción de los factores esenciales para el crecimiento neuronal.
- Apoptosis: muerte prematura de las neuronas por un mecanismo de "suicidio celular".
- Factores genéticos: la llamada ELA familiar incluye entre el 5% y 10% de los casos diagnosticados.
Cuadro clínico
Es muy variado y suele empezar con una sensación de fatiga, contracciones musculares (fasciculaciones), calambres y torpeza en alguna extremidad. Otras veces puede afectar el habla, o producir espasticidad de las extremidades. Las manos y pies son las partes del cuerpo que se afectan de manera temprana por la debilidad muscular, muy pronto aparecen dificultades en la marcha y en otras actividades cotidianas, como bañarse o vestirse. La debilidad, paresia o parálisis puede extenderse a músculos del cuello y tronco, produciendo problemas al tragar, masticar o respirar. Otras veces puede haber espasticidad generalizada, con risas y llantos inapropiados, que son parte de la enfermedad y no se deben a una alteración mental. La ELA sólo ataca las neuronas motoras. Sin embargo, las neuronas de los núcleos oculomotores no se ven afectadas, ni tampoco la vista, oído, olfato, gusto y tacto. De ninguna manera se afecta el esfínter de la vejiga (núcleo de Onuff) o el recto. Las funciones musculares autonómicas de corazón, intestino y líbido permanecen intactas. Se estima que más del 50% de las motoneuronas se pierden antes de que los síntomas sean evidentes.7
Signos y síntomas
Se relacionan con la región en donde se localiza la afección de las motoneuronas (Tabla 1).

La evolución de la ELA corresponde a una debilidad progresiva, con un rango amplio de discapacidades asociadas. Algunas veces son afectados todos los músculos bajo control voluntario y se pierde la fuerza (principalmente de miembros superiores), capacidad de mover brazos, piernas y finalmente todo el cuerpo. Cuando fallan los músculos del diafragma y de la pared torácica se pierde la capacidad de respirar sin la asistencia de un ventilador mecánico. La mayoría de los pacientes con ELA muere por falla respiratoria y neumonía (aunque puede ser por arritmia cardíaca y broncoaspiración), generalmente entre dos y cinco años después del inicio de los síntomas. En los fallecimientos debidos a falla respiratoria se ha encontrado además atrofia en las raíces nerviosas cervicales y torácicas.8 El pronóstico de la enfermedad es poco alentador, para el tercer año después del diagnóstico 50% de los pacientes con ELA fallecen,2 mientras que solo un 10% alcanza una sobrevida de 10 años o más.1 La notable excepción del famoso físico Stephen Hawking, quien lleva alrededor de 40 años con el diagnóstico de ELA, probablemente se debe a que padece la variedad que ha sido denominada "ELA Benigna" o a que padezca otro tipo de enfermedad de neurona motora. Este término fue utilizado por primera vez por Forbes Norris, un neurólogo americano, para referirse a pacientes con ELA que han mantenido estable su sintomatología en los últimos nueve meses o han tenido una lenta progresión de su enfermedad. Este término no es ampliamente aceptado entre los especialistas. Después de la revisión de la literatura médica actual se puede concluir que la enfermedad es relativamente antigua, con alrededor de 140 años de haber sido descrita, siendo destacable el mal pronóstico de la misma, aunado a las muchas formas de tratamiento empleadas sin resultados favorables.
Diagnóstico
Se basa en una historia clínica detallada, un examen físico completo, estudios de conducción nerviosa, estudios de laboratorio y de imagen, ya que no existe una prueba que confirme un diagnóstico definitivo de ELA, aunque la presencia de señales de deterioro de las NMS y NMI en una sola extremidad constituye una fuerte sospecha.
El diagnóstico de ELA está basado principalmente en los síntomas y signos observados en el paciente, además de una serie de pruebas que descartan otras enfermedades. Sumado a una historia clínica completa, se lleva a cabo un examen neurológico minucioso a intervalos regulares para evaluar si los síntomas como la debilidad muscular, atrofia muscular, hiperreflexia y espasticidad empeoran progresivamente.4,7
Una herramienta clínica importante para evaluar la debilidad muscular es la electromiografía, la cual debe ser realizada en los cuatro miembros, el tronco y el cuello. Para este objetivo pueden emplearse las escalas de Kendall.9
Es claro entonces que el diagnóstico de ELA es básicamente de exclusión, descartando enfermedades psiquiátricas, oncológicas o metabólicas, lo cual justifica la realización de diversos estudios de imagen, laboratorio y electroconducción, entre otros.
Pruebas auxiliares del diagnóstico
- Electromiografía (EMG): estudio que evalúa la función de nervios y músculos. Constituye la prueba fundamental para el diagnóstico de afección de la NMI.
- Exámenes de sangre: para descartar afecciones metabólicas como hiperparatiroidismo, tirotoxicosis, hiperinsulinismo, entre otros.
- Pruebas de función pulmonar: con especial énfasis en la determinación de la capacidad vital forzada (CVF), para establecer si los músculos respiratorios se encuentran afectados.
- Resonancia Magnética o Tomografía Computarizada de la columna cervical: para verificar la ausencia de enfermedades como siringomielia, mielopatía cervical espondilótica, tumores o malformaciones vasculares medulares.
- Pruebas genéticas: si existen antecedentes familiares de ELA, para descartar enfermedades como la adrenoleucodistrofia, deficiencia de hexosaminidasa A, etc.
- Estudios del mecanismo de la deglución.
- Punción lumbar: para descartar enfermedades como la de Guillain-Barré.
- Biopsia de músculo estriado: para excluir miositis por cuerpos de inclusión o miopatía distal.
Criterios actuales de diagnóstico
Las últimas modificaciones al diagnóstico de ELA se realizaron en el año de 1998, en una reunión en la ciudad de Arlie, Texas, USA. De acuerdo a estos criterios se necesita cumplir con lo siguiente para realizar el diagnóstico de ELA.3
1. Signos de compromiso de NMI (por examen clínico, electrofisiológico y/o neuropatológico), en una o más de cuatro regiones medulares (bulbar, cervical, torácica o lumbosacra) (Tabla 1).
2. Signos de compromiso de NMS (por examen clínico), en una o más de estas cuatro regiones (Tabla 1).
3. Progresión de los signos hacia otras regiones a lo largo del tiempo.
Además, la escala de Escorial, mencionada anteriormente,10 incluye lo siguiente:
1. Ausencia de evidencia electrofisiológica o patológica de otra enfermedad o proceso que pueda explicar los signos de degeneración de NMS y NMI.
2. Ausencia de evidencia en los estudios de neuroimagen de otro proceso o enfermedad que pueda explicar los signos clínicos y electrofisiológicos.
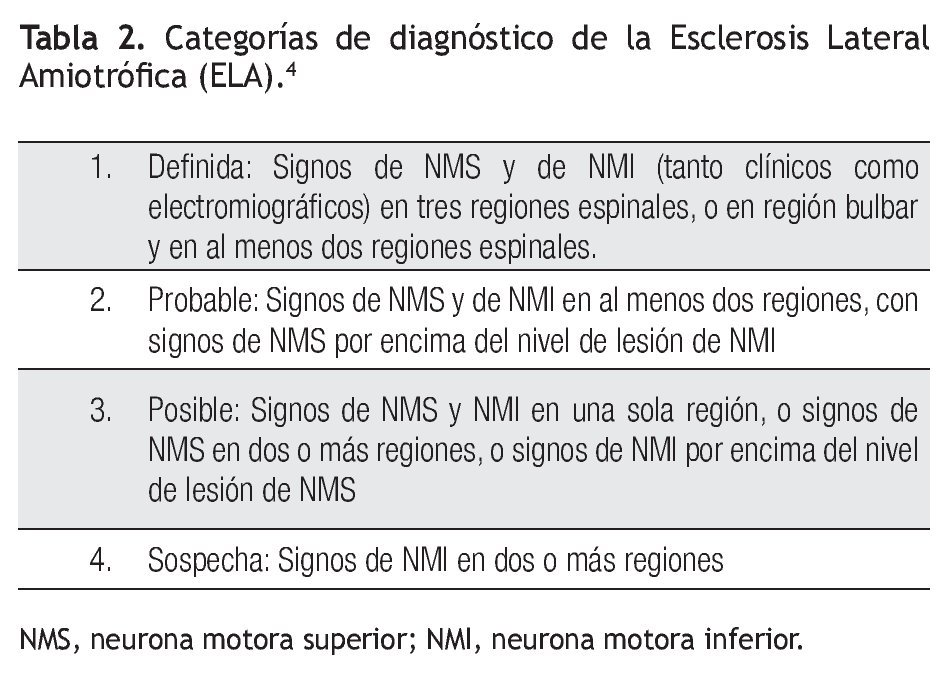
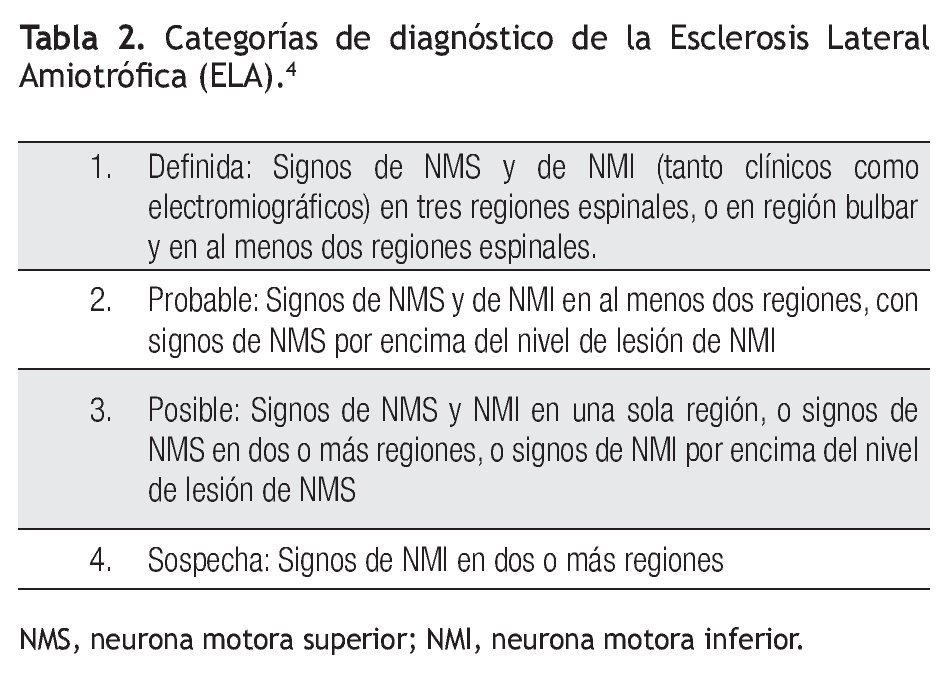
De la localización y combinación de los signos de NMI y NMS en las cuatro regiones citadas se establecieron los niveles de certeza diagnostica de ELA: a) definida, b) probable, c) posible, d) sospecha. Esta clasificación resulta poco clara, por lo cual es difícil aplicarla universalmente. Las categorías se describen en la Tabla 2.
Para el seguimiento de la evolución de los pacientes con ELA se utiliza la Escala de Clasificación Funcional de la Esclerosis Lateral Amiotrófica (The Amyotrophic Lateral Sclerosis Functional Rating Scale revised, ALSFRSr),11 que evalúa diez funciones, entre ellas el habla, salivación, masticación, escritura, manejo de utensilios, vestido e higiene, marcha, subir escaleras, respiración y movilización en cama.
Tratamiento
Actualmente se encuentra limitado a la terapia física, respiratoria (mediante uso de Biphasic Positive Airway Pressure, BIPAP), ocupacional y del habla, que pueden contribuir a la mejoría en las funciones de la vida diaria.
El tratamiento farmacológico está dirigido a aliviar la sintomatología, principalmente empleando fármacos para disminuir la espasticidad, los calambres, los trastornos de la deglución, la sialorrea, la insuficiencia respiratoria y las alteraciones emocionales.4
Desde 1996 se encuentra en el mercado el Riluzole en presentación de tabletas de 50 mg. Este medicamento actúa inhibiendo los procesos metabólicos en los cuales interviene el glutamato, el neurotransmisor excitador que tiene relación con la etiopatología de la ELA. Su mecanismo de acción es mediado por el bloqueo de los canales de Na, que disminuye el influjo de iones de Ca+2, lo que previene la estimulación de los receptores de glutamato. El Riluzole se administra a una dosis de 50mg cada 12 horas por vía oral, con dosis mayores no se han observado cambios significativos en la mejoría clínica, por lo cual se prefiere la dosis de 100 mg por día.12
Entre los efectos benéficos del tratamiento con este compuesto se ha visto que la sobrevida aumenta entre tres y cinco meses, también disminuye la necesidad de ventilación mecánica, lo cual representa un incremento en la mejoría de un 10% a 15% en comparación con otras formas de terapia. Este fármaco no ha demostrado tener un papel destacado en mejorar la función motora, pulmonar o la aparición de fasciculaciones. Los costos del tratamiento con Riluzole son cercanos a los $3 500 pesos mexicanos mensuales, en Inglaterra se refiere un costo de 5£ por cada dosis de 50mg. A lo anterior debe sumarse el costo de la terapia física y los demás costos asociados en el largo plazo.
Recientemente se reportó un estudio de Riluzole combinado con carbonato de litio, el cual incluyó 16 pacientes; el grupo control (n=28) estaba tratado exclusivamente con Riluzole, la sobrevida a 18 meses en el grupo que recibió el tratamiento combinado fue del 100%, mientras que en los pacientes del grupo control alcanzó 71%. La heterogeneidad clínica de los pacientes incluidos no permitió establecer si existía una diferencia estadísticamente significativa.13
Terapia celular intratecal de la ELA
En diversos modelos animales se ha observado una mejoría significativa en respuesta a esta forma de terapia, con un aumento en la glía y la neuroplasticidad, además de mejoría en la función motora. También se refiere mejoría clínica al aplicar G-CSF (factor estimulador de colonias de granulocitos) antes de la infusión de células progenitoras autólogas, además de sugerir la aplicación de dosis múltiples de estas infusiones de células madre autólogas, a intervalos de tres meses, debido a que éstas viven alrededor de 12 a 15 semanas, lo que favorecería un resultado positivo con el trasplante. Se ha encontrado, tanto en humanos como en modelos animales, que la dosis óptima de infusión intratecal que conduce a mejores resultados clínicos es de 1x106 células hematoprogenitoras CD34+, habiéndose comparado en estudios previos con dosis de 1 x 104 y 1 x 105.14,16
Entre las diversas vías de administración de hematoprogenitores autólogos obtenidos de la médula ósea se incluyen la intraparenquimatosa, intravenosa e intratecal. Un estudio realizado en Ankara, Turquía, incluyó 13 pacientes, 10 hombres y tres mujeres, con un rango de edad entre los 34 y 71 años. El tratamiento consistió en realizar una laminectomía a nivel de C1-C2, posteriormente se inyectaron las células madre en el trayecto de las astas anteriores de la médula espinal. Los cuatro sitios inyectados fueron el parénquima cerebral (10 millones de células), la duramadre (cinco millones), el espacio subaracnoideo (cinco millones) y por vía intravenosa (cinco millones), posteriormente se realizó un seguimiento por un año. Nueve pacientes mejoraron notablemente, uno permaneció estable y tres fallecieron a causa de infecciones pulmonares e infarto agudo al miocardio.17
Un estudio realizado en Monterrey, Nuevo León, México, en el año 2009, incluyó 20 pacientes con ELA definida, 10 pacientes sometidos a terapia celular intraparenquimatosa y 10 con ELA definida manteniéndose en tratamiento convencional como grupo control. La población incluida en el protocolo fue de cinco mujeres y cinco hombres, con un rango de edad entre 38 y 62 años. El tratamiento consistió en aplicar una dosis subcutánea de 300 microgramos de G-CSF por tres días consecutivos, para posteriormente recolectar las células mesenquimales CD133+ por leucoféresis. A cada paciente se le aplicó una dosis de 2.5 - 7.5 x 105 CD133+ células madre que se inyectaron a una profundidad de 7mm en la corteza cerebral (franja motora definida por esterotaxia o neuronavegación). Los pacientes permanecieron en observación un día, y egresaron sin sufrir efectos adversos. Se concluyó que la terapia celular intraparenquimatosa es segura, factible y bien tolerada en los pacientes con ELA, además de que puede ofrecer la posibilidad de detener la progresión de esta enfermedad.18
En cuanto a la vía de administración intratecal, en la literatura médica actual se encuentran reportados cinco estudios experimentales en humanos, los cuales incluyen en total a 31 pacientes con ELA tratados con terapia celular intratecal. Del total de 31 pacientes, seis (19%) respondieron a esta forma de tratamiento, la mejoría observada fue un aumento de la fuerza en dos pacientes (6.45%) y cuatro pacientes aumentaron su CVF (12.9%). Cabe destacar que en ninguno de estos estudios se reportaron efectos secundarios asociados a la inyección intratecal.19-22 A septiembre del 2011, en el sitio Clinical Trials están registrados tres estudios experimentales de tratamiento de ELA con terapia celular autóloga intratecal, empleando dosis de 1 x 106 células mononucleares (CMN) CD34+. Estos se están desarrollando en Murcia, España, la Clínica Mayo en Minnesota, USA y la Universidad de Hanyang, en Corea del Sur. El número de pacientes incluidos en estos centros va desde uno, en Minnesota, hasta 71 en Corea, a la fecha no se han publicado los resultados de ninguno de los tres estudios, aunque se espera que estén terminados para finales del año 2012.23-26
Con estos datos se puede concluir que la terapia celular intratecal con células progenitoras autólogas ha sido estudiada de manera insuficiente y sin resultados concluyentes, por lo que estudios adicionales en pacientes con ELA de reciente diagnóstico son necesarios para definir el papel preciso de esta potencial estrategia de tratamiento en esta catastrófica enfermedad.
Conclusiones y perspectivas
La ELA es una enfermedad neurodegenerativa, cuyo origen y tratamiento eficaz permanecen por definir. Aunque la enfermedad es infrecuente, también es cierto que es devastadora, de tal manera que los esfuerzos por encontrar una cura o un tratamiento que lleve a una mejor calidad y duración de la vida continúan, habiéndose incluso seleccionado el día 21 de Junio como el "Día Mundial de la ELA", desde 1997.27 De las distintas formas de terapia estudiadas, la farmacológica ha producido escasa mejoría, por lo que es previsible que las diferentes variedades de terapia celular en esta enfermedad, ya sea por vía intravenosa, intraparenquimatosa (intracerebral en franja motora o intraespinal en asta anterior), o intratecal, seguirán siendo exploradas hasta obtener datos definitivos al respecto de la eficacia de cada una de ellas. Finalmente, es importante mencionar que la terapia génica de la ELA se encuentra en fase de desarrollo en modelos animales, aunque sin resultados concluyentes, por lo que todavía es prematuro asignarle un papel en el manejo de ELA en seres humanos.28
Correspondencia: Dr. José Carlos Jaime Pérez.
Servicio de Hematología, Edificio "Dr. Rodrigo Barragán", 2° piso, Hospital Universitario "Dr. José Eleuterio González" Avenida Madero y Gonzalitos S/N, Colonia Mitras Centro, Monterrey, Nuevo León, C.P. 64460. México.
Teléfono/Fax: (52) (81) 1257 2905 y 1257 2906.
Correo electrónico: carjaime@hotmail.com.
Recibido: Noviembre 2011.
Aceptado: Noviembre 2011