


La enfermedad de von Hippel-Lindau (VHL) es un síndrome de neoplasias de herencia autosómica dominante, que predispone a lesiones en sistema nervioso central (SNC) y vísceras. las neoplasias de origen primario o metástasico que afectan SNC, representan la condición más frecuente que simula ictus apopléjico.
Se presenta mujer de 38 años de edad, con signos y síntomas de focalización neurológica en fosa posterior de inicio agudo, evidenciándose en resonancia magnética hemangioblastoma cerebeloso asociado a enfermedad de VHL, simulando ictus apopléjico.
La presencia de signos y síntomas de lesión en fosa posterior de inicio agudo en un paciente joven, debe hacer sospechar que su causa es de probable naturaleza hemorrágica, por lo que algunos autores han reportado que: "Todo ictus apopléjico que en su inicio se acompañe de crisis hipertensiva es un ictus hemorrágico intraparenquimatoso hasta demostrar lo contrario". no obstante, en este caso la presencia de cefalea y cifras de presión arterial normales, coincidieron con neoplasia intracerebral simulando ictus apopléjico debida a enfermedad de VHL.
Von Hippel-Lindau disease (VHL) is an autosomal-dominant neoplasia syndrome, pre-disposed to develop lesions of central nervous system (CNS) and viscera. A primary or metastatic neoplasia affecting CNS represents the most common condition that mimics stroke.
A 38-year-old woman with neurologic signs and symptoms in posterior fossa of acute onset, magnetic resonance imaging demonstrated cerebellar hemangioblastoma associated with VHL disease mimicking stroke.
The presence of signs and symptoms of injury in posterior fossa of acute onset in a young patient must arise suspicion of probable hemorrhagic nature, As some authors have stated: "All stroke that begins accompanied with hypertensive crisis, is an hemorrhagic intraparenchymal stroke until shown otherwise". Nevertheless in this case, headache and normal arterial pressure coincided with a neoplasia simulating stroke due to VHL disease.
Introducción
la enfermedad de von Hippel-Lindau (VHL) es un síndrome de neoplasias de herencia autosómica dominante (prevalencia de 1 en 39.000 nacidos vivos), que resulta de una mutación germinal del gen supresor de tumores VHL localizado en 3p25.5, relacionado con neoplasias localizadas en el sistema nervioso central (SNC) y vísceras.1 La proteína de VHL es directamente responsable de la regulación en baja de la familia del factor inducible por hipoxia (FIH), que controla múltiples factores de transcripción asociados con el desarrollo de carcinomas de células renales y hemangioblastomas.2
El diagnóstico clínico requiere al menos una manifestación característica de VHL, en el contexto de historia familiar de la enfermedad. sin embargo, en ausencia de historia familiar positiva para VHL, la presencia de al menos dos hemangioblastomas en SNC; o hemangioblastoma único en combinación con neoplasia visceral típica, constituyen criterios diagnósticos establecidos.3 Cabe señalar, que a pesar de no poseer manifestaciones cutáneas observadas en las facomatosis, la enfermedad de VHL se considera entre este grupo de afecciones.4
Los hemangioblastomas son neoplasias benignas de histogénesis incierta clasificada como grado 1 según la organización Mundial de la salud (oMs), que representan alrededor del 1% a 2% de las neoplasias del SNC y del 7% a 10% de las neoplasias primarias de fosa posterior.5 Usualmente se presentan en adultos jóvenes y se localizan predominantemente en cerebelo, aunque pueden observarse en tallo encefálico, hemisferios cerebrales y cordón medular. Entre 10% a 40% de los hemangioblastomas cerebelosos se diagnostican en pacientes con VHL, en quienes el 70% de los hemangioblastomas se localizan en cerebelo.4,6 las neoplasias viscerales en VHL incluyen carcinomas y quistes, localizados en riñón, páncreas y órganos anexos reproductores.1
Las neoplasias de origen primario o metástasico que afectan SNC, representan la condición más frecuente que simula ictus apopléjico, seguida de las malformaciones vasculares, hematoma subdural y enfermedades infecciosas.7 no obstante, las neoplasias localizadas en SNC pueden manifestarse como síndrome de hipertensión endocraneana, primoconvulsión o enfermedad mental orgánica. sin embargo, en el contexto de signos o síntomas de focalización neurológica de inicio agudo, una neoplasia es capaz de imitar clínicamente un ictus apopléjico inherente a su localización anatómica.8
Exponemos el caso de una paciente que cumple con criterios clínicos y radiológicos de VHL simulando ictus de fosa posterior.
Presentación del caso
Paciente femenina de 38 años de edad, que acude al servicio de Emergencia del Hospital Universitario de Maracaibo, por presentar cefalea, vértigo y lateralización de la marcha a la izquierda, de inicio agudo. niega antecedentes de hipertensión arterial sistémica, diabetes mellitus o historia familiar de neoplasias u otras enfermedades de índole neurológica. Refiere intervención quirúrgica en julio de 2008 debida a quistes ováricos bilaterales de naturaleza benigna; no obstante, señaló ser habitualmente sana. Dos meses antes de la enfermedad actual, se efectuó cesárea segmentaría de su tercer gesta, niega abortos. En el ingreso, presentó presión arterial de 110/70 mmHg y frecuencia cardiaca de 72 latidos/min.
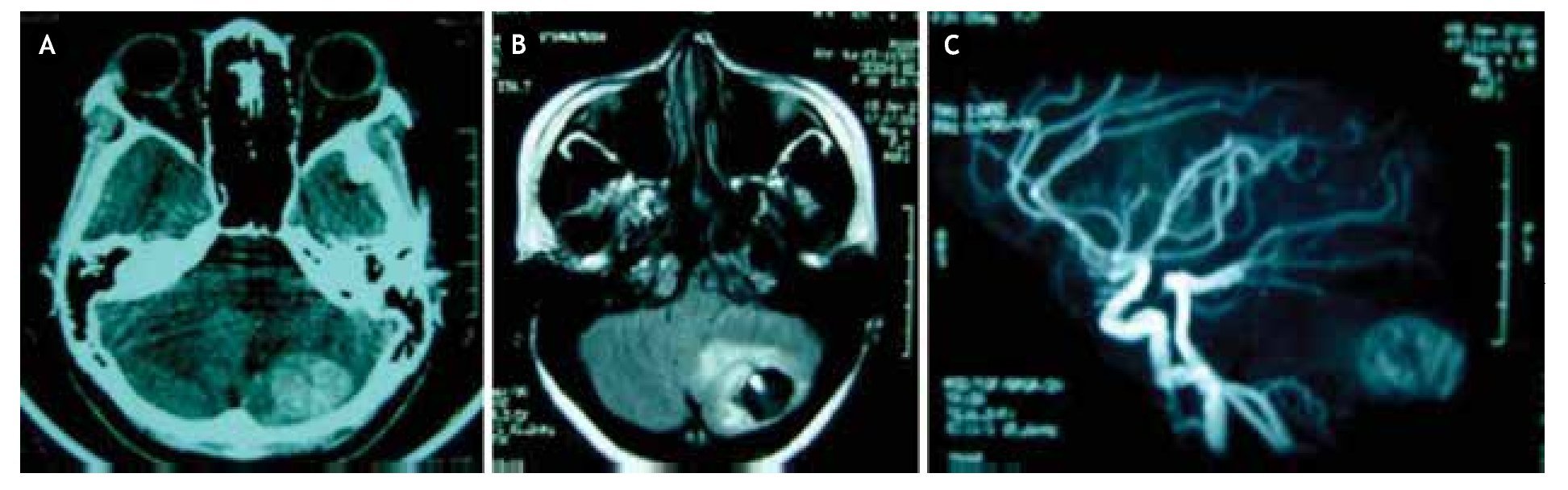
Al examen neurológico, la paciente se encontraba consciente, orientada en persona, tiempo y espacio, pupilas reactivas e isocóricas con fondo de ojo normal, sin alteraciones en la sensibilidad o pares craneales, fuerza muscular 5/5 global; observándose objetivamente lateralización de la marcha a la izquierda y reflejo patelar pendular izquierdo, el resto de las pruebas de exploración cerebelosa fueron normales. no presentó rigidez de nuca. A la admisión, ingresa con el diagnóstico clínico de ictus apopléjico isquémico del hemisferio cerebeloso izquierdo. Posteriormente, se realiza tomografía axial computarizada (TAC) cerebral simple, que demostró imagen hiperdensa heterogénea en el hemisferio cerebeloso izquierdo. Por ende, se solicita resonancia magnética cerebral (RMC) con gadolinio y fase vascular, evidenciando imagen quística con nódulo hiperintenso en su interior y ovillo vascular compatible con hemangioblastoma (Figura 1). se realizó ecograma pélvico, que reportó la presencia de múltiples quistes bilaterales en órganos anexos reproductores (ligamentos anchos), corroborados con tomografía de abdomen y pelvis. Asimismo, se determinaron concentraciones elevadas de marcador CA-125 (174 UI/ml), hemoglobina y hematocrito normales (12,3 g/dl y 35%, respectivamente), sin anormalidad en el resto de los estudios complementarios.
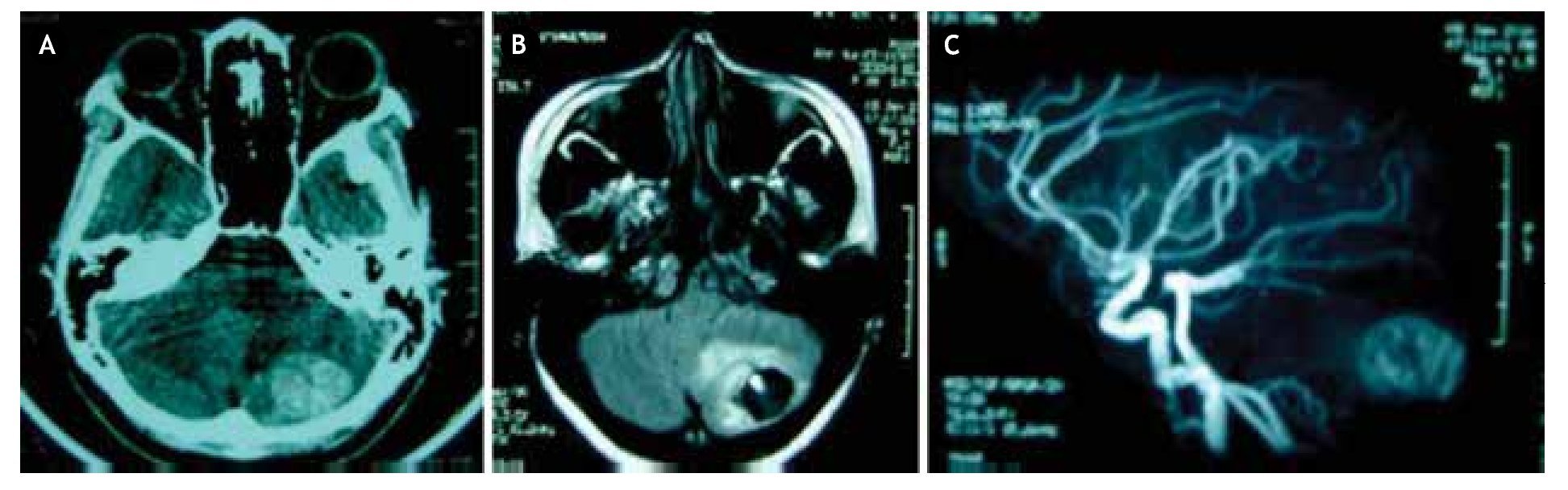
Figura 1. neuroimágenes de hemangioblastoma en paciente con enfermedad de von Hippel-Lindau. A) En la tomografía simple, se observa imagen hiperdensa heterogénea en hemisferio cerebeloso izquierdo. B) En resonancia magnética con secuencia potenciada en T1 plano axial, se observa lesión de aspecto quístico con nódulo hiperintenso en su interior y edema perilesional, confirmando en fase vascular, C) la presencia de neoplasia extensamente vascularizada típica del hemangioblastoma, en hemisferio cerebeloso izquierdo.
La paciente evolucionó satisfactoriamente en 72 horas, por lo cual fue dada de alta tras una semana de hospitalización, egresando con diagnóstico clínico y radiológico de enfermedad de VHL. En la actualidad, se encuentra en control por los servicios de neurología y neurocirugía.
Discusión
Los hallazgos radiológicos en nuestra paciente son consistentes con los criterios diagnósticos clínicos de VHL, establecidos por Melmon y Rosen en 1964.9 En consecuencia, ante la ausencia de antecedentes familiares de VHL, la presencia de un hemangioblastoma y una lesión visceral son concluyentes para apoyar el diagnóstico.4
En pacientes con hemangioblastomas de SNC y diagnóstico de enfermedad de VHL, se presentan síntomas neurológicos en una edad más temprana con respecto a los casos esporádicos de hemangioblastoma.10 El promedio de edad para el diagnóstico de hemangioblastomas cerebelosos asociado con la enfermedad de VHL es de 30 años, observándose en algunas casuísticas una edad media al diagnóstico de 39 años.11,12 En general, existe un pico de incidencia entre los 35 a 45 años, siendo excepcionales tanto en el anciano como en la infancia.6,12
En los casos de pacientes con hemangioblastoma cerebeloso, la tríada de cefalea, vómitos y síntomas cerebelosos de evolución subaguda o crónica, constituyen las características clínicas más comunes en su presentación, siendo el inicio agudo categóricamente inusual.5,6,13 Cabe destacar, que se trata de tumores de crecimiento muy lento que clínicamente se manifiestan por obstrucción en la circulación del líquido cefalorraquídeo provocando hipertensión endocraneana con presencia de papiledema y signos de disfunción cerebelosa como vértigo, ataxia, hipotonía, dismetría y adiadococinesia.6
En nuestro caso, previo a comprobar la presencia radiológica de una neoplasia en fosa posterior, la presentación clínica de cefalea, vértigo y lateralización de la marcha de inicio agudo, sugirió un fenómeno de naturaleza vascular cerebral.
Tradicionalmente, los ictus se han clasificado por su naturaleza en isquémicos y hemorrágicos, considerando básicamente para el diagnóstico diferencial el perfil temporal de los síntomas y signos neurológicos.14 Usualmente, el ictus isquémico no suele acompañarse de cefalea y nunca el paciente presenta rigidez de nuca. En el análisis clínico, si ocurre cefalea y signos de focalización neurológica de inicio súbito, siempre precedidos de elevación de las cifras de presión arterial, es el ictus apopléjico de naturaleza hemorrágica la presentación más frecuente.
Anatomopatológicamente, la mayoría de las hemorragias intraparenquimatosas relacionadas con hipertensión arterial se localizan en o cerca de la bifurcación de las pequeñas arterias penetrantes (50-700 micras de diámetro) de la arteria basilar o arterias cerebral media, anterior o posterior. las ramas de las pequeñas arterias, a menudo tienen múltiples puntos de ruptura, y algunos están asociados con necrosis fibrinoide del subendotelio y capas de agregados de plaquetas y fibrina.14,15 Estas lesiones se caracterizan por la rotura de la lámina elástica, atrofia y fragmentación del músculo liso, disección y degeneración celular de tipo granular o vesicular.15 Estos sucesos permiten deducir que los vasos debilitados en su pared deben romperse por una presión intraluminal incrementada, y que razonablemente obedece a una crisis hipertensiva.14
En nuestro caso, aunque la paciente presentó síntomas y signos de focalización neurológica en fosa posterior de inicio agudo, con presencia de cefalea, las cifras de presión arterial normales, minimizaron la probabilidad de ictus apopléjico de naturaleza hemorrágica. El hallazgo por imágenes (hemangioblastoma cerebeloso y quistes bilaterales en órganos anexos reproductores) y exámenes complementarios entre los que destaca el marcador tumoral CA-125 elevado, determinaron la presencia de neoplasia, simulando ictus apopléjico debida a enfermedad de VHL. la ausencia de policitemia en nuestro caso fue considerada, aún cuando esta última es observada sólo entre 5% a 20% de los casos de pacientes con enfermedad de VHL.4
Las teorías de neoplasias simulando ictus apopléjico comprenden mecanismos que circunscriben un aumento repentino en la proporción de edema, asociado con la lesión y el efecto de masa, que origina compromiso del flujo sanguíneo e isquemia.7 Asimismo, la hemorragia intratumoral podría dar lugar a déficits neurológicos de inicio brusco, mostrando signos de hemorragia en la neuroimagen, lo que hace probable esta posibilidad como mecanismo causal en nuestra paciente.
Las cuidadosas observaciones y correlaciones clínico-patológicas de Eugen von Hippel y Arvid lindau han proporcionado la descripción de una entidad clínicogenética, reconocida como una importante enfermedad predisponente a neoplasias hereditarias y que llevó al descubrimiento de un gen clave en regulaciones fisiológicas significativas.16 El gen VHL se expresa ampliamente en los tejidos, incluso en tejidos no afectados por la enfermedad y codifica la proteína de VHL (pVHL). Postraduccionalmente, la pVHL forma complejos con elogina B, elongina C, Rbx1 y Cullin 2, constituyendo una ligasa de ubiquitina que proteolisa la subunidad α del FIH.1,17
En circunstancias normales, el FIH coordina la respuesta celular a la hipoxia por medio de la regulación transcripcional, que aumenta el metabolismo celular y amplía la expresión de factores angiogénicos y mitogénicos.18 Algunos de estos factores, incluyen el factor de crecimiento vascular endotelial (VEGF), la cadenaβ del factor de crecimiento derivado de las plaquetas (PDGF-β), la eritropoyetina y el factor transformador de crecimiento beta.1,18
En la enfermedad de VHL la ausencia o alteración de la pVHL, condiciona que el FIH pueda estimular constitutivamente la angiogénesis por medio del aumento de las concentraciones de VEGF o PDGF-β, explicando la naturaleza vascular de las neoplasias asociadas con el fenotipo clínico. Además, el aumento en la permeabilidad vascular que es mediada por VEGF es la causa de la formación frecuente de edema peritumoral y quistes en la enfermedad de VHL.1,18
Por lo tanto, dado la naturaleza vascular del hemangioblastoma y el elevado flujo sanguíneo que recibe, es razonable que la primera manifestación de la lesión pueda ser sangrado espontáneo,4 explicando el inicio súbito de manifestaciones neurológicas inherentes a la localización anatómica de la lesión.
El seguimiento clínico e imagenológico de los pacientes con enfermedad de VHL es pertinente en intervalos de uno a dos años, aún cuando se mantengan asintomáticos, permitiendo identificar más de 75% de nuevas lesiones, algunas de las cuales pueden requerir intervención quirúrgica como en el caso de nuestra paciente.5
En la actualidad, las causas más comunes de muerte en pacientes con enfermedad de VHL son los carcinomas de células renales y complicaciones neurológicas por hemangioblastomas cerebelosos.19 Junto con la existencia de los nuevos enfoques que brindan los estudios moleculares del gen de VHL, deben mantenerse los méritos y ventajas de la visión integral, dada por el análisis clínico y patológico de esta enfermedad.
En conclusión, la presencia de signos y síntomas de lesión en fosa posterior de inicio agudo en un paciente joven, debe hacer sospechar que su causa es de probable naturaleza hemorrágica. Esta consideración es sustentada en la observación clínica desde 1980 cuando se inicia el primer curso de postgrado de la Escuela neurológica del Zulia, que nos permitió enunciar el siguiente postulado: "Todo ictus que en su inicio se acompañe de crisis hipertensiva es un ictus hemorrágico intraparenquimatoso hasta demostrar lo contrario".14 no obstante, en este caso la presencia de cefalea y cifras de presión arterial normal en la paciente, coincidió con el hallazgo de neoplasia simulando ictus apopléjico debida a enfermedad de VHL.
Conflicto de intereses
Los autores declaran no tener conflicto de intereses.
Financiamiento
Los autores no recibieron ningún patrocinio para llevar a cabo este artículo.
Correspondencia:
Dr. Carlos J. Chávez.
Instituto de Investigaciones Biológicas. Facultad de Medicina, Universidad del Zulia, Apartado 526,
C.P. 4001, Maracaibo, Venezuela.
Teléfonos: (+58) 261 7597250. Fax: (+58) 261 7597249.
Correo electrónico: biomolecula@hotmail.com
Recibido: Agosto 2012.
Aceptado: octubre 2012