


Introducción
Las neoplasias de células plasmáticas son un grupo de entidades caracterizadas por la proliferación de una clona maligna de células, habitualmente productoras de inmunoglobulinas, que pueden presentarse como lesión única (plasmocitoma solitario) o como múltiples lesiones (mieloma múltiple).1-2 Los plasmocitomas solitarios (PS) constituyen una enfermedad muy poco frecuente, representan 5% a 8% de las enfermedades de las células plasmáticas. La enfermedad se presenta predominantemente en hombres entre 55 y 65 años de edad.3,4 La localización más común del plasmocitoma solitario es el hueso, en cuyo caso la enfermedad se denomina plasmocitoma óseo (PO), correspondiendo a 50% a 70% de los casos. El resto de los plasmocitomas solitarios se localizan en tejidos blandos, los cuales se conocen como plasmocitomas extra-medulares (PE).2,5-6 Los huesos más frecuentemente involucrados son aquellos que cuentan con hematopoyesis activa; por orden descendente de frecuencia: vértebras torácicas, lumbosacras, cervicales, pelvis, costillas, extremidades superiores, cara, cráneo y fémur. La mayoría de los PE se localizan en cabeza y cuello, más frecuentemente en la vía aerodigestiva superior.2,5 El tratamiento de elección es la radioterapia localizada, con dosis de 40 a 50 Gy.2,5-11 El uso de quimioterapia adyuvante es controvertido. Aproximadamente 50% a 60% de los PO y 10% a 20% de los PE progresarán a MM.2,5
En el presente estudio se describe la frecuencia, formas de presentación y evolución de los pacientes con diagnóstico de PS atendidos en una institución universitaria.
Métodos
Se realizó una búsqueda en los archivos del Servicio de Anatomía Patológica y el Servicio de Hematología del Hospital Universitario Dr. José E. González, en el periodo de tiempo entre enero de 2003 y diciembre de 2010, para identificar pacientes con diagnóstico histopatológico de plasmocitoma. Se revisaron los expedientes clínicos para incluir los siguientes datos: género, edad, signos y síntomas iniciales, tamaño y localización del plasmocitoma, hemoglobina, creatinina, calcio, albúmina, globulinas, β-2 microglobulina, inmunoelectroforesis de proteínas séricas, número de células plasmáticas en médula ósea, estudio de imagen realizado, tratamiento, fecha de progresión, de muerte o de última visita.
Fueron identificados 37 casos con diagnóstico de plasmocitoma. Dieciséis fueron excluidos, cinco por no contar con expediente clínico, 10 porque los estudios de extensión revelaron la presencia de enfermedad sistémica siendo clasificados como MM y un paciente que se encontraba en tratamiento por MM de ocho años de evolución en quien el diagnóstico de plasmocitoma fue la manifestación de progresión de la enfermedad. Veintiún pacientes cumplieron criterios para ser clasificados como plasmocitoma solitario: biopsia del tumor con la presencia de células plasmáticas clonales; sin evidencia de plasmocitomas en otros sitios con estudios de imagen, ya sea radiografía (serie ósea metastásica), resonancia magnética nuclear (RMN) o tomografía computarizada (TC); aspirado o biopsia de médula ósea con <10% de células plasmáticas, además de ausencia de anemia, hipercalcemia o insuficiencia renal atribuibles a la discrasia de células plasmáticas.2,12 Para la definición de respuesta completa, parcial y progresión a MM se siguieron los lineamientos internacionales.2 La supervivencia libre de enfermedad se analizó con el método de Kaplan-Meier utilizando el programa SPSS versión 17.
Resultados
En 21 pacientes con diagnóstico histopatológico de plasmocitoma, los estudios de extensión descartaron mieloma múltiple y fueron clasificados como PS, 11 (52%) hombres y 10 (48%) mujeres. La edad mediana de presentación fue 57 años (rango 25 a 80). Cuatro pacientes (19%) eran menores de 50 años.
Dieciséis pacientes (76%) presentaron PO y cinco (24%) pacientes PE. El síntoma inicial más frecuente fue dolor en 13 pacientes (62%), 11 pacientes con PO manifestaron dolor óseo y dos pacientes con PE localizado en pleura y estómago iniciaron con dolor pleurítico y abdominal, respectivamente.
Al momento del diagnóstico, 13 pacientes contaban con TC, tres con RMN y cinco sólo tenían radiografía simple. En 16 casos se realizó biopsia incisional del tumor, en dos biopsia excisional de la masa, en un caso se realizó biopsia por aspiración con aguja fina y en dos casos no fue posible determinar el método utilizado para obtención de la muestra para estudio histopatológico.
Los 21 pacientes contaban con aspirado de médula ósea realizado por punción unilateral de cresta iliaca, en todos los casos el porcentaje de células plasmáticas fue menor de 10%, en cuatro casos el porcentaje se encontraba entre 5% a 9%, en 11 pacientes <5% y en seis casos el informe sólo indicaba <10% de células plasmáticas.
Dieciséis pacientes contaban con electroforesis de proteínas séricas, de los cuales dos (12%) tenían presencia de patrón monoclonal; once contaban con la medición de β2-microglobulina, en dos pacientes fue mayor de 3.5 mg/L. En 10 casos tuvimos información del tamaño del plasmocitoma por medio del estudio de imagen; en tres, el diámetro fue mayor a 5 cm. La dosis de radioterapia varió entre 30 Gy y 45 Gy. Los esquemas de quimioterapia consistieron en ciclofosfamida, dexametasona y talidomida; prednisona y melfalán o dexametasona y talidomida.
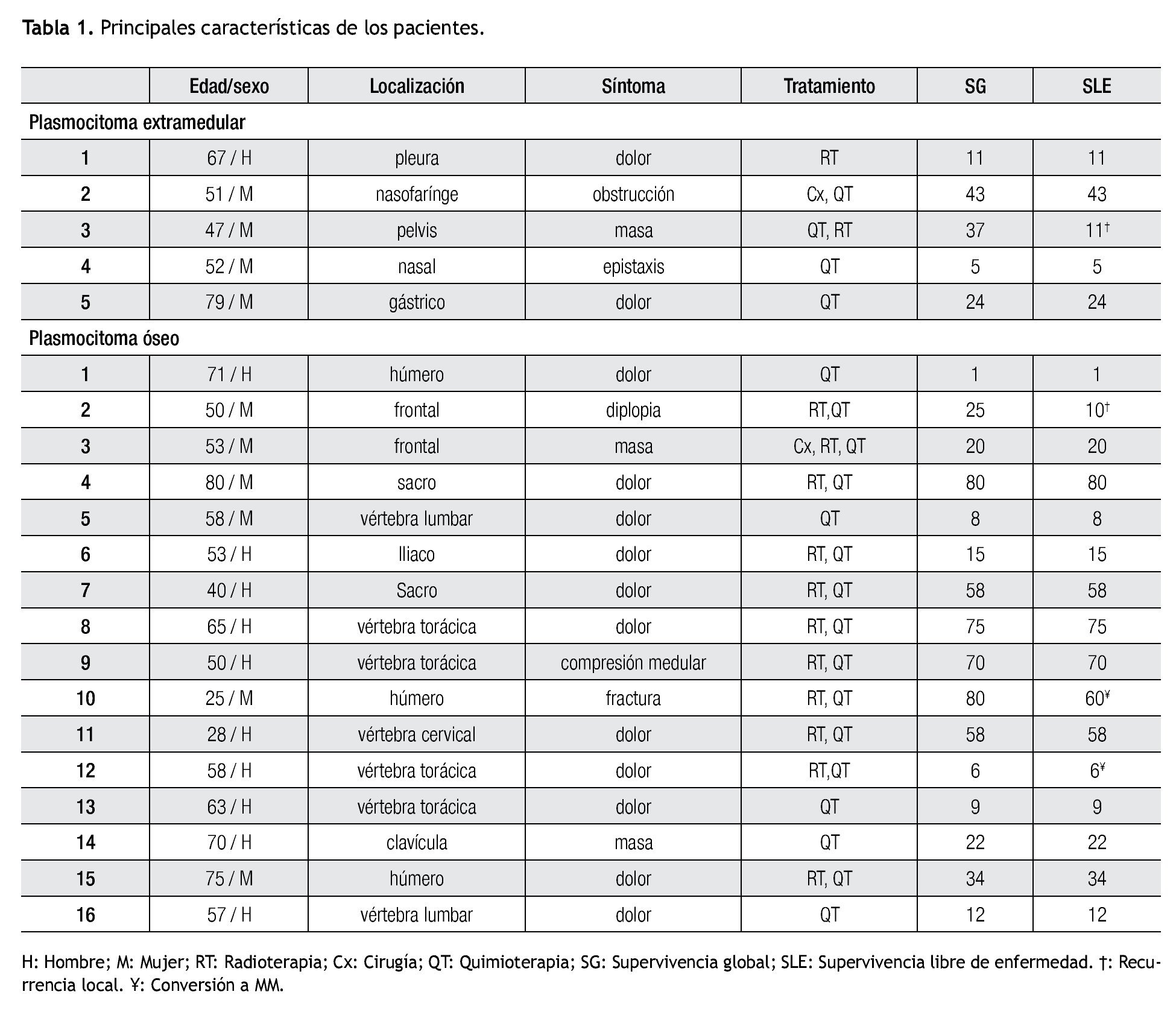
Plasmocitoma extra-medular. De los cinco pacientes con diagnóstico de PE, fueron cuatro mujeres, la edad mediana de presentación fue 52 años (rango 47 a 79). En dos pacientes el PE se localizaba en vías aéreas superiores (Tabla 1). Cuatro pacientes fueron tratados con quimioterapia, en dos pacientes como monoterapia y en dos pacientes combinada, uno con radioterapia y otro como adyuvante posterior a cirugía. Un paciente recibió sólo radioterapia con 40 Gy en 20 sesiones. El tiempo de duración de la quimioterapia varió de tres a seis meses. La mediana de seguimiento fue de 24 (cinco a 43) meses. Ningún paciente con PE ha desarrollado mieloma múltiple hasta la fecha del último seguimiento. Cuatro casos lograron respuesta completa. La paciente con plasmocitoma en hueco pélvico, tratado con quimio y radioterapia, presentó aumento del tamaño del plasmocitoma sin evidencia de enfermedad sistémica después de 11 meses de seguimiento. La supervivencia libre de enfermedad a tres años fue de 75% (Figura 1).
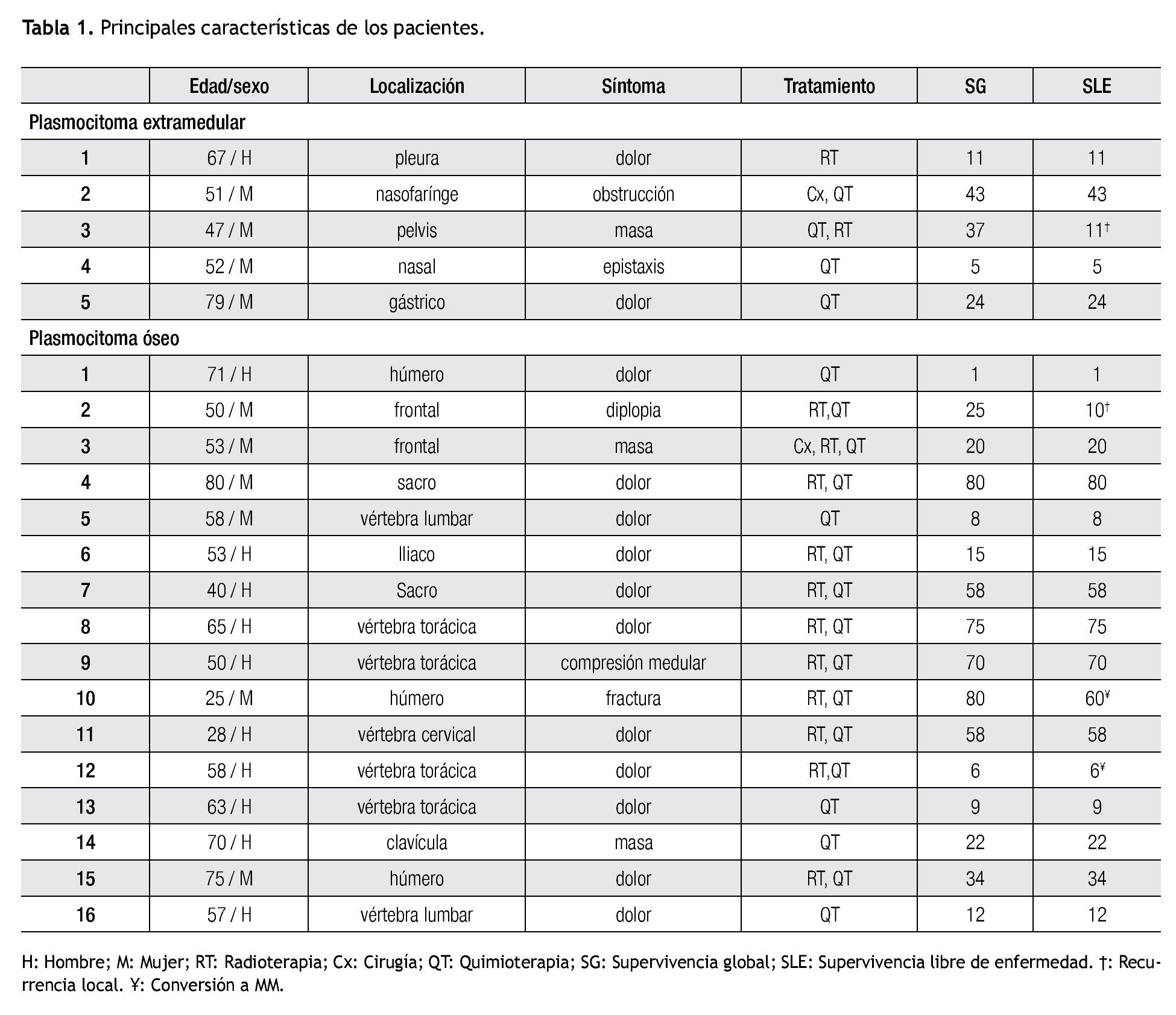
Figura 1. Supervivencia libre de enfermedad.
Plasmocitoma óseo. De los 16 pacientes con diagnóstico de PO, 10 (62%) fueron varones, la mediana de edad fue 57 años (rango 25 a 80). La localización más frecuente fue en vértebras toracolumbares en seis casos (38%), tres (19%) en húmero, dos (13%) en sacro, dos (13%) en hueso frontal (Tabla 1). El dolor óseo fue el síntoma inicial y el motivo de consulta en 11 casos (69%). La modalidad de tratamiento más frecuente utilizada fue la combinación de radioterapia y quimioterapia en 10 pacientes, en un paciente se realizó cirugía para excisión completa del plasmocitoma seguida de radioterapia y quimioterapia, cinco pacientes recibieron sólo quimioterapia.
Con una mediana de seguimiento de 23 meses (rango 1 a 80) un paciente desarrolló recurrencia local del PS y dos pacientes (12%) han progresado a MM. Una mujer de 50 años con diagnóstico de PO frontal con diplopía como síntoma inicial, recibió tratamiento con quimioterapia por 8 meses y radioterapia 45 Gy, desarrolló recurrencia local a los 10 meses del diagnóstico. De los dos pacientes que progresaron a MM, un paciente de 58 años con diagnóstico de PO en vértebra torácica, tratado con radioterapia 30Gy y quimioterapia por cuatro meses progresó a MM seis meses después del diagnóstico y una paciente que desarrolló MM cinco años después del diagnóstico de PO del húmero a los 25 años, manifestado por una fractura patológica y tratado inicialmente con radioterapia seguido de quimioterapia seis meses. La supervivencia libre de enfermedad a cinco años en el grupo de pacientes con PO fue de 64% (Figura 1).
Discusión
El plasmocitoma es una proliferación clonal de células plasmáticas las cuales son idénticas por morfología e inmunofenotipo a las observadas en el MM pero que se manifiestan de manera localizada ya sea en tejido óseo o blando. La razón por la que un paciente desarrolla PS o MM no está clara, pero se sabe que está relacionada con las moléculas de adhesión así como a la expresión de receptores de citocinas de las células plasmáticas malignas.13
En el presente estudio la proporción de mujeres con diagnóstico de PS, y en particular de PE fue mayor a la descrita en la literatura. La modalidad de tratamiento más utilizada fue quimioterapia. Con una mediana de seguimiento de 24 (11 a 80) meses, cuatro pacientes (19%) desarrollaron recurrencia local o sistémica.
El PS similar a otras enfermedades de las células plasmáticas es más frecuente en varones, estudios previos han encontrado una relación masculino-femenino cercana a 3:1.3-5,11,14-15 Datos que difieren de nuestra población de estudio en la cual ambos géneros presentan una frecuencia similar siendo de 48% mujeres y 52% varones. Notablemente cuatro de los cinco casos de PE fueron mujeres.
La recomendación internacional indica que el tratamiento de elección del PS es la radioterapia localizada, con dosis de 40 Gy a 50 Gy2,7-10 seguida de vigilancia. La dosis de radiación ha sido establecida con base en estudios retrospectivos, Mendenhall y colaboradores encontraron que 31% de los pacientes que recibían menos de 40 Gy presentaban falla local al tratamiento comparado con 6% de quienes recibían dosis mayores.16 Franssica y colaboradores no observaron fallas al tratamiento utilizando 45 Gy o más.11 Dos pacientes desarrollaron recurrencia local del PS a los 10 y 11 meses, ambos recibieron quimioterapia inicial seguida de radioterapia 35 y 45Gy.
Aproximadamente 50% a 60% de los PO y 10% a 20% de los PE progresan a MM, con una mediana de tiempo para progresión de 34 (rango 13 a 180) meses.5,9,15-17 Los estudios que apoyan el uso de quimioterapia adyuvante muestran que es posible retrasar o prevenir la conversión del PO a MM y mejorar la calidad de vida del paciente.8,15,18 Uno de estos estudios fue realizado en la Ciudad de México de forma prospectiva con una mediana de seguimiento de 8.9 años, la conversión a MM fue de 54% en el grupo que recibió sólo radioterapia y 12% en quienes recibieron radioterapia seguida de quimioterapia, es importante resaltar que el esquema administrado fue melfalán con prednisona cada seis semanas por tres años.18 En otras series de pacientes la quimioterapia adyuvante no ha mostrado que evite la progresión o mejore la supervivencia, por lo que estos estudios sugieren tratar con radioterapia localizada y observar para iniciar tratamiento sistémico al momento de desarrollar la progresión a MM.14,19-20 Dos de nuestros pacientes progresaron a MM a los seis y 60 meses del diagnóstico, ambos recibieron quimioterapia adyuvante por seis y nueve meses.
Existen algunos factores que predicen un mayor riesgo de conversión a MM: tamaño >4cm, localización en vértebras, persistencia de la proteína monoclonal en suero, edad y β2-microglobulina elevada.5 De nuestros dos pacientes que progresaron a MM, en uno el PS se localizaba en vértebra torácica, el otro tenía diámetro mayor de 5 cm y ninguno tenía proteína monoclonal detectable en suero al diagnóstico. Ambos recibieron tratamiento combinado con quimioterapia y radioterapia.
El diagnóstico y clasificación de las enfermedades de las células plasmáticas depende en gran medida del juicio clínico al seleccionar las herramientas de estudio ya que los avances tecnológicos han permitido aumentar la precisión diagnóstica. Es probable que el grupo de pacientes con diagnóstico de PS que progresa a MM en corto tiempo, tenga enfermedad diseminada desde el inicio y por lo tanto mayor riesgo de desarrollar MM. La RMN puede detectar enfermedad previamente no sospechada en pacientes con diagnóstico de PS hasta en 26% a 30% de los casos.21 La citometría de flujo y la detección molecular de cadenas ligeras identifica células plasmáticas clonales en la médula ósea de pacientes que no tienen evidencia de infiltración al análisis microscópico. Además, la tomografía por emisión de positrones (PET), de reciente introducción, es útil para detectar enfermedad oculta hasta en 35% de los pacientes con aparente PS.22
En el presente estudio, 13 pacientes contaban con TC y sólo tres con RMN inicial, en ningún caso se realizó PET, la evaluación de la médula ósea para cuantificar células plasmáticas se realizó por análisis microscópico. Es probable que con el uso del PET la incidencia del PS disminuya y que aumente el número de respuestas completas así como el índice de curación de los pacientes con verdadero PS.
La conducta terapéutica inicial en nuestro centro incluye radioterapia y quimioterapia, en casos seleccionados se omite la radioterapia con la intención de no retrasar el tratamiento sistémico, que habitualmente es bien tolerado, o cuando es alto el riesgo desarrollar complicaciones asociadas a la radiación. Otra razón importante para decidir evitar la radioterapia, principalmente en casos de plasmocitoma en zona lumbar y pélvica es la inevitable afección a la médula ósea, lo que en algunos casos impediría la posibilidad de tratamiento con quimioterapia así como de un trasplante de médula ósea en el futuro.
Aunque la presente serie de casos es pequeña y el seguimiento corto, la progresión a MM fue menor a la descrita en la literatura, lo que coincide con los estudios en que la quimioterapia adyuvante disminuye o al menos retrasa la progresión a MM. Sin embargo, la recurrencia local fue mayor a la esperada.
Conclusión
Consideramos necesario estudios comparativos utilizando criterios diagnósticos más estrictos para definir si la quimioterapia adyuvante pudiera beneficiar a un grupo de pacientes PS.
Correspondencia: Dra. Luz del Carmen Tarín Arzaga.
Av. Madero esquina con Av. Gonzalitos s/n. Col. Mitras Centro. C.P. 64460.
Teléfono: 83488510, fax: 86756717.
Correo electrónico:tarinarzaga@prodigy.net.mx
Recibido: octubre 2011.
Aceptado: octubre 2011