


Obesity has become a public health problem around the world. According to the Organisation for Economic Co-operation and Development (OECD, 2014 report), more than one in three adults in Mexico are obese. It is known that the hypothalamus, a region of the Central Nervous System (CNS), is actively involved in regulating energy homeostasis during obesity. Anatomically, the hypothalamus is composed of several nuclei coordinating body weight and metabolism, including the arcuate nucleus (ARC), which contains neurons co-expressing orexigenic peptides like Agouti-related protein (AgRP), Neuropeptide Y (NPY) and the anorexigenic peptide Pro-opiomelanocortin (POMC). During obesity, the integration and metabolic response in the ARC is disrupted by three molecular mechanisms: (1) activation of endoplasmic reticulum (ER) stress, (2) mitochondrial dysfunction, and (3) increase of ER and mitochondria contacts, known as Mitochondria-Associated Membranes (MAMs). In this context, it is proposed that MAMs formation induces mitochondrial Ca2+ overload and metabolic dysfunction, leading to insulin resistance and diabetes. Recently, MAMs formation has emerged as one of the molecular mechanisms underlying metabolic alterations during obesity. Thus, in this review we will focus on proposing scientific evidence to support the role of the MAMs and their function on calcium regulation during obesity, as an important pathological mechanism in the development of diabetes mellitus type 2.
Obesity represents one of the diseases with the highest prevalence worldwide. The World Health Organization estimated that in 2014 about 13% of the adult population was obese and the amount of obesity in individuals has doubled since the 1980s.1 In Mexico, the Organization for Economic Co-operation and Development (OECD) reported that approximately one in three adults suffers from obesity,2 considering a body mass index (BMI) higher than 30kg/m2 to be obese. Usually, an increase in BMI is linked to the excessive intake of high caloric foods, or a decrease in catabolism.1–3 However, this variable seems to be multifactorial, involving changes in behavior, metabolic, environmental, genetic, cultural and socioeconomic factors. Obesity increases the possibility of developing cardiovascular diseases, hypertension, type 2 diabetes, dyslipidemias, some types of cancer, arteriosclerosis and cognitive deterioration, affecting the patient's quality of life,1,3,4 thus making the understanding of the cellular and physiological mechanisms implicated in this pathology relevant for the development of new treatment approaches.
Compelling scientific evidence suggests that the central nervous system (CNS), and especially the hypothalamus, are key in the control of energy homeostasis because of their effects over of food intake regulation, lipid accumulation in the body and body weight. Therefore, defects in metabolic integration involving insulin, leptin and ghrelin, at a hypothalamus level, generate alterations in energy homeostasis related to the development of obesity and health comorbidities.5 The toxic effect associated with the increase of different types of lipids, such as saturated fatty acids (SFA), triglycerides and ceramides in obesity is called lipotoxicity.7–9 In this context, lipids are known to induce a failure in the endoplasmic reticulum (ER) and mitochondria functions, promoting oxidative stress in response to an unfolded protein response (UPR) exacerbation10–12 as well as the development of type 2 diabetes and metabolic syndrome.6,12–16
At a mitochondrial-associated-membrane level (MAM), obesity regulates the communication between ER and the mitochondria. In this microdomain, obesity coordinates the recruitment of proteins in MAMs as well as the Ca2+ flow between ER and mitochondria at the hypothalamus and liver level.17–19
In this review, we will discuss the role of the hypothalamus in the regulation of energy consumption and its effect on obesity and diabetes development in the context of MAMs remodeling, alterations in Ca2+ homeostasis, activation of ER stress and mitochondrial function.
The hypothalamus is a brain region which is key in the control of energy homeostasisThe hypothalamus is part of the diencephalon, located under the thalamus and formed by different nuclei. Prominent among these are the preoptic, paraventricular, suprachiasmatic, lateral, posterior, mammillary, dorsomedial (DMH), ventromedial (VMH) and arcuate (ARC). As a whole, the activation of the different nuclei regulates energy homeostasis through their action over food intake, thermogenesis, and glucose balance in plasma, among other things.20 In particular, DMH, VMH, ARC, the medial portion of the lateral hypothalamus and the ventral premammillary nucleus are related to the regulation of food intake. Therefore, they are called the “satiety center”, while the lateral hypothalamus represents part of the “feeding center”.20–22,25 All these hypothalamic nuclei are part of an integral system called the melanocortin system which modulates with the regulation of energy and satiety.
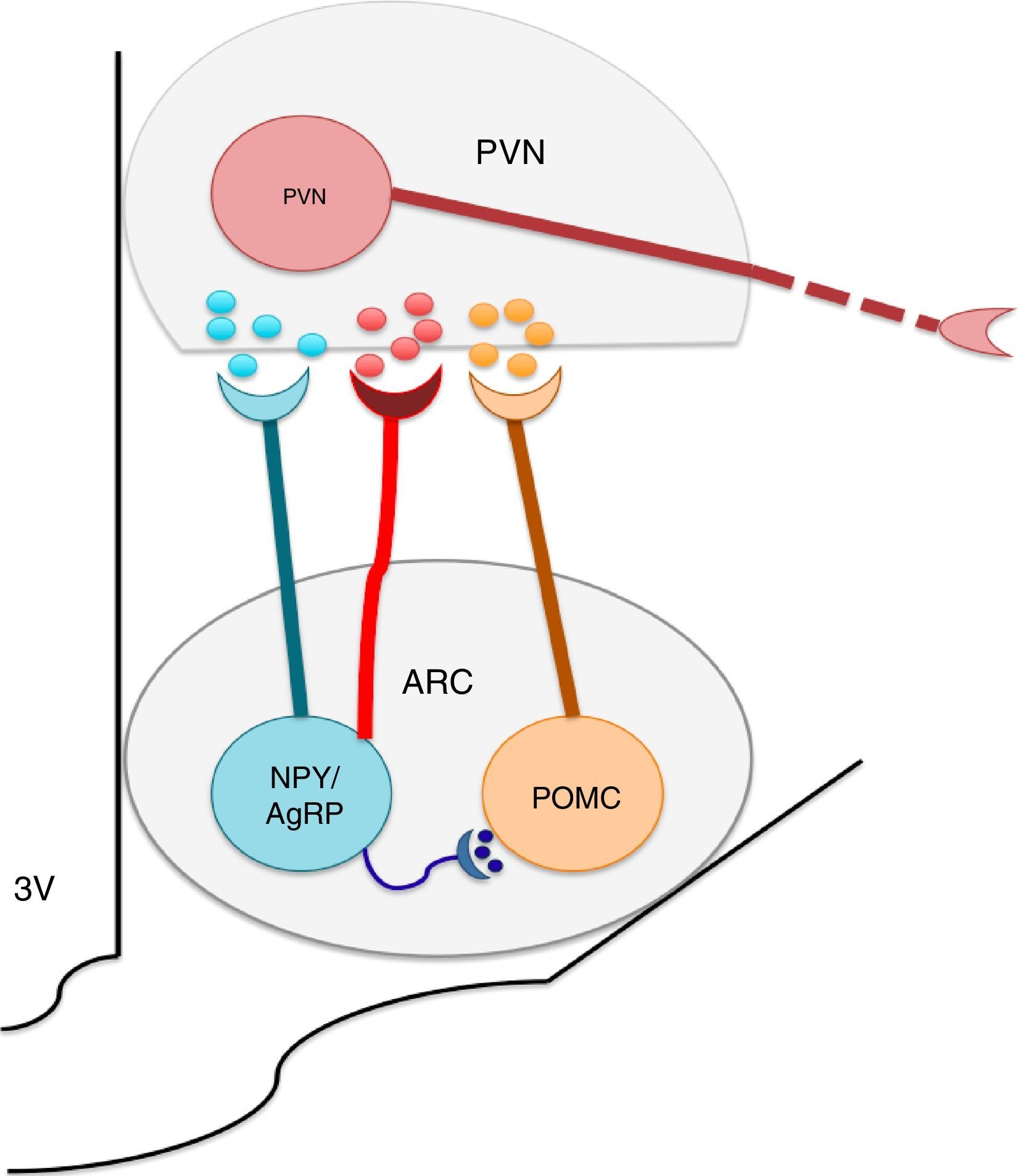
The nuclei which form a part of the melanocortin system possess selective specificity for synthesis and the liberation of neuropeptides and neurotransmitters according to their cellular type identity. Thus, ARC is formed by orexigenic neurons which express the Neuropeptide Y (NPY)/the agouti related peptide (AgRP), anorexigenic neurons, and proopiomelanocortin (POMC), which release POMC. The latter is processed into a melanocytes α-stimulating hormone (αMSH). NPY/AgRP and POMC/αMSH increase and decrease respectively, in appetite.22–24 It is known that the greatest concentrations of neurons expressing insulin receptors is located at the ARC level. ARC and its fibers project to the paraventricular nucleus and periformical region (Fig. 1).23 Arc is found in a privileged position within the hypothalamus since its proximity to the median eminence (ME) allows it to sense hormonal and biochemical factors of blood and cerebrospinal fluid with the purpose of regulating energy homeostasis,22,23,25 among which leptin, insulin and ghrelin stand out. Leptin and insulin, secreted by adipocytes and the pancreas respectively, produce anorexigenic effects through their action over ARC, producing leading to an increase in the genetic expression of NPY/AgRP and a decrease of POMC in ARC,23,24,26,27,29,30 while ghrelin, secreted by the stomach, promotes food intake31,32 through the stimulation of the GHSR1 receptor in ARC's NPY/AgRP neurons, inhibiting its activity through the action of the gamma aminobutyric acid (GABA) neurotransmitter.33–35
 Neuropeptide Y/Agouti related-proteins (NPY/AgRP) and propiomelacortics (POMC) coordinate the activity of the paraventricular (PVN) nucleus, promoting the stimulation or inhibition of apetite, respectively. GABA is a neurotransmitter released by the NPY/AgRP neurons, and is very important to the control of food intake, inhibiting the anorectic effects of the POMC neurons.")
The arcuate nucleus neurons (ARC) Neuropeptide Y/Agouti related-proteins (NPY/AgRP) and propiomelacortics (POMC) coordinate the activity of the paraventricular (PVN) nucleus, promoting the stimulation or inhibition of apetite, respectively. GABA is a neurotransmitter released by the NPY/AgRP neurons, and is very important to the control of food intake, inhibiting the anorectic effects of the POMC neurons.
In animal models of obesity, induced by high-fat diet intake or in obese patients, the presence of insulin and leptin resistance has been demonstrated. Insulin resistance is a key factor in the development of type 2 diabetes mellitus (DMT2).36 Physiologically, the effects of insulin resistance are known to be manifested in the pancreas, muscle and hypothalamus. Glucose homeostasis in the body is maintained due to the action of insulin on adipocytes and muscle. At first, the pancreas β cells release insulin, which binds to its receptor on the adipocytes and hepatocytes, and promotes the translocation of vesicles from the cytoplasm to the plasmatic membrane containing GLUT-4 transporter.37 Molecularly, insulin attaches to the α subunit of the insulin receptor (IR), inducing the activation of the tyrosine kinase domain in the intracellular portion of the β subunit38 and the phosphorylation of three tyrosine residues (Tyr-1158, Tyr-1162 and Tyr-1163).38 The signal is amplified through the downstream phosphorylation insulin receptor substrate type proteins (IRS-1, IRS-2, IRS-3, IRS-4), recruiting Shc, growth factor 2 (Grb2/SOS) and regulatory subunit p85 of the inositol phosphatidiyl 3′-kinase (PI2 kinase).38–42 Amongst all these, PI3K plays a major role for the activation of the Akt/PKB and PKCζ cascade. Once activated, the Akt protein induces glycogen synthesis through the inhibition of the glycogen synthase kinase protein (GSK-3), the synthesis of mTOR (mammalian target of rampamycin) core protein and cellular survival by the inhibition of the apoptosis.39–42 However, in insulin-resistant conditions, such as the ones presented during lipids increase in obesity, phosphorylation is inhibited in IRS1 tyrosine and increasing 307 serine phosphorylation of IRS1.37,43 Pancreatic cells respond by upregulating the synthesis and liberation of insulin leading to β cells dysfunction and DMT2 development.44 On the other hand, obesity also induces insulin resistance in the hypothalamus, where the decrease in the number of receptors to this hormone is observed.45,46 Altogether, hyperglycemia during obesity is linked to alterations in: (1) decrease in tyrosine phosphorylation, (2) decrease in the number of receptors to insulin, and (3) increase in the plasmatic levels of insulin. Furthermore, an increase in blood leptin concentrations has been demonstrated as a leptin-resistance index in obese patients and obesity-induced murine models by high-fat diet intake.28 Leptin signalling pathway is activated by leptin binding to its receptor and the downstream activation of Janus (JAK) kinases by transducers and activators of transcriptions (STAT), MAPK and IRS1.47,48 In the context of obesity, leptin resistance is linked to the reduced activity of the transductor and the phosphorylated activator of transcription 3 (STAT)48,49 and to the no-activation of genes of the cytokine suppressors family (SOCS).49 In fact, it has been recently demonstrated that leptin and insulin act as a whole on hypothalamic neurons,50 suggesting that obesity affects hypothalamic circuits and compromises the hormonal integration implicated in the coordination of energetic metabolism.
Additionally, the effect of these hormones on body physiology not only relates to obesity problems, but also to the development of hypertension in mice and humans51–53 and Alzheimer's susceptibility.54 Thus, the mechanisms which underlie the functional alterations of the hypothalamus during obesity might be implicated in additional pathologies in humans.
Alterations in the endoplasmic reticulum and mitochondria function during obesity promote insulin and leptin resistanceDifferent studies have provided substantial evidence that obesity induces endoplasmic reticulum (ER) stress and mitochondrial dysfunction into organs which are metabolically relevant for the coordination of energetic balance. ER is a network of membranous tubes and flattened sacs which is present in all eukaryotic cells, involving synthesis, folding, maturation and traffic of proteins, as well as storage and homeostasis of Ca+2 and lipid biosynthesis.12,15,55,56 Proteins enter into the ER lumen as unfolded polypeptide chains and they fold in the interior to make a more stable formation. When the proteins are misfolded, they overload, and the expression of chaperones is activated with the inability to control its accumulation, with the objective of stimulating the unfolded protein response (UPR). Protein synthesis, and its translocation toward ER, are reduced in this process, and UPR genes are activated to increase the ability of ER to fix misfolded proteins. If homeostasis cannot be reestablished, a mechanism of cell death by apoptosis is stimulated.12,15,57 This regulation mechanism is very active and especially important in secreting cells41 such as neurons.11
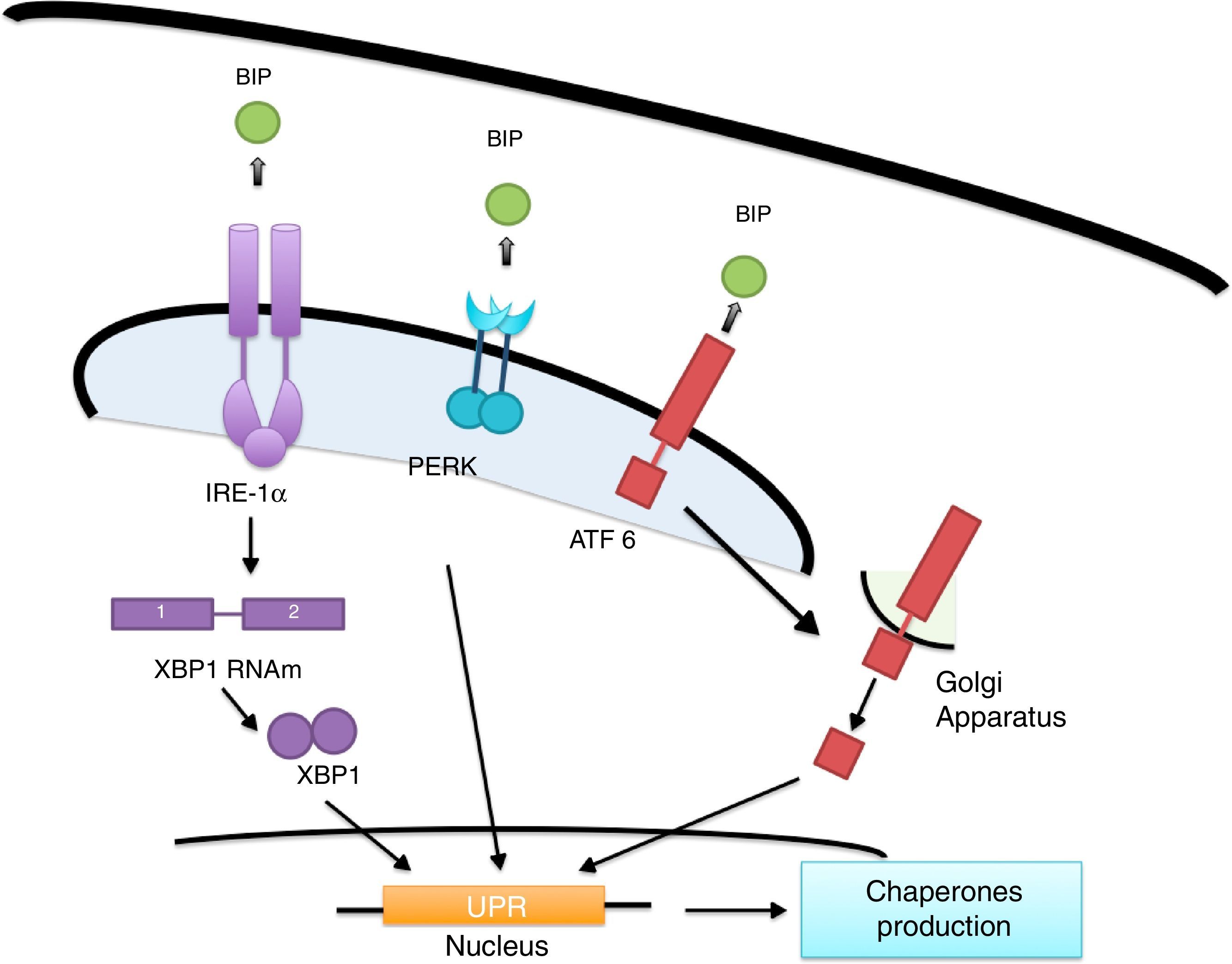
ER's stress sensors and UPR onsetThe increase of misfolded proteins in ER is well-known to produce stress, which may be measured by select membrane protein groups called: IRE, PERK and ATF6.58,59 Ire1α, as well as Ire1β, break the ARNm of the X-box binding protein (XBP1) generating XBP1 splicing (sXBP1).59 sXBP1 activates the expression of genes which codify for the synthesis of chaperones, components of the degradation of ER associated with protein degradation (ERAD) and genes in charge of phospholipid biosynthesis and autophagy.60–62 The PERK complex phosphorylates the translation initiation factor eIF2a, dimming translation, yet boosting ATF4 translation, which regulates genes with secretor functions and of the apoptotic type included in the C/EBP homologous (CHOP) protein.60,62 ATF6 is an activator factor which travels to the Golgi apparatus in response to stress, where it undergoes proteolysis, liberating its cytosolic domain, called the bZip transcription factor, which enters the nucleus and activates the UPR genes involved in protein folding, processing and degrading.60 The chaperones which regulate the proper protein folding are the proteins related to GRP78 glucose, also known as heavy chain binding immunoglobulin proteins,58 protein disulfide-isomerase (PDI), calnexin (CANX), calreticulin (CALR) and GRP94.59 In this context, BiP binds to Ire1, ATF6 and PERK domains and dissociates after ER stress induction, suggesting that unfolded proteins compete for BiP, allowing the oligomerization and activation of these sensors (Fig. 2).51,60,61
Lipid accumulation during obesity creates ER stress inducing hypothalamic inflammation and the generation of diabetes. When the flow of misfolded proteins to the ER is increased, the BIP protein chaperone is released from IRE-1α, PERK and ATF6, allowing it to act as a transcription factor and allowing chaperone proteins synthesis, which improve protein folding.")
Epidemiological data confirms a strong link between the increase in the level of obesity and the development of type 2 diabetes; indicating that for every kilogram of gained weight, at the population level, there is a linear increase in the diabetes rate.3 Experimental evidence, in obese humans and animal models with obesity, suggests that the escape of lipids from adipose tissue and ectopic accumulation of ceramides (a type of sphingolipid), acylcarnitines, diacylglycerols and AGS cause tissue damage to metabolically relevant organs, including the skeletal muscle, liver, pancreatic beta cells, myocardium and brain, in an event called lipotoxicity.63–65 The lipotoxic effect is largely determined because every organ has its own lipid profile. Hence, selective changes in lipid species in different organs may be relevant to the development of lipotoxicity. In this context, it is known that, physiologically, C18:0 type ceramides are essential for cerebellar development and C22:0-24:0 regulates hepatic function,63,64 while saturated diacylglycerols and lipids take part in intracellular signaling processes in many cellular types of the body.65 In this regard, it has been suggested that during obesity new species of lipids, which are potentially toxic for the body's organs, are produced, including ceramides, cholesterol, AGS and diacylglycerols. All these species are known to inhibit insulin sensitivity in cellular cultures and animal models.66 Saturated ceramide and lipid accumulation has even been detected in the skeletal muscle of obese humans, which correlates to a generation of insulin resistance.66,67 Of note, ceramides, cholesterol, saturated DAG and lipids activate the ER's stress in the hypothalamus in animal models, generating insulin resistance.9,11,66,68
Notably, a close connection is known to exist between the ER stress activation and inflammation. Experimental work shows that Jun-N terminal kinase (JNK activating protein 1 – AP1-), activated during the ER's stress promotes the activity of the IKKB/NF-κB pathway.69 De Sousa et al. (2005), proved that high fat diet-induced obesity increases the TNFα levels and other pro-inflammatory cytokines in serum of animal models of obesity.45 In contrast with unsaturated lipids, intra-cerebrovascular infusion of saturated lipids and palmitic acid cause inflammation and insulin resistance, which is a IKKβ-kinase dependent pro-inflammatory,70 presumably through the activation of TLR4 recetors.71,72 In this context, the increase of SFA, cholesterol, triglycerides, DAGs and ceramides during obesity causes the activation of ER stress and inflammation, inducing the insulin resistance state in the hypothalamus and generating metabolic complications.
Lipotoxicity linked to lipid increase during obesity produces mitochondrial dysfunction, insulin resistance and the development of diabetesMitochondria represents pivotal intracellular organelles in charge of ATP synthesis through oxidation of energetic substrates and cellular processes, including cell differentiation and cellular death, and help maintain growth control and the cellular cycle.73 Defects in mitochondrial function leads to a metabolic deterioration increasing susceptibility to the development of type II diabetes mellitus or obesity.74
Different studies on the muscle of obese humans with a insulin resistance state have shown that the presence of mitochondrial dysfunction, including low oxidative enzymatic activity and a decrease in lipid metabolism compared to thin subjects.75 Furthermore, SFA increase during obesity correlates with primary defects of the mitochondrial oxidative capacity of the muscle, liver and adipose tissue, promoting DAG increase and insulin resistance.76 These events are closely linked with the increase in T2DM.73,77 A big part of the lipotoxic effect of lipids over the mitochondrial function is because its accumulation produces reactive oxygen species (ROS), including anion superoxide (O2−), hydrogen peroxide (H2O2) and hydroxyl radical (OH−).73 A fatty dietary intake in mice has been proven to increase ROS production in the liver and adipocytes and precedes TNFα increase in blood and liver.78 A similar effect has been found in the hypothalamus of rats with acute hypertriglyceridemia, caused by intraperitoneal or intra-cerebrovascular injections of 20% lipids including a combination of soy oil, glycerin and phospholipids.79 In this context, we were able to demonstrated the lipotoxic neuronal damage at metabolic center regulator caused by the injections of lipids and promoting the consumption of food.79 This effect seems to be linked to ROS increase in the POMC neurons of the hypothalamus, which produce satiety, hence the suggestion that POMC neurons are more sensitive to lipotoxic damage caused by ROS compared to NPY/AgRP neurons. Conversely to these events, the administration of complex antioxidants prevents ROS formation and inhibits appetite, in part by inducing the expression of POMC in ARC.80 Consequently, mitochondrial dysfunction linked to the development of obesity modulates food intake and sensitivity to insulin, coordinating the development of T2DM.
By now, the cause of mitochondrial damage during obesity and its effect on insulin sensitivity is unknown. In fact, mitochondrial dysfunction is proposed to be a consequence and not a cause of insulin. This is based on the fact that alterations in the mitochondrial function in diverse models do not decrease insulin sensitivity.81 Even the lipid infusion, which promotes insulin resistance, increases beta oxidation in humans and, contrarily, an increase in insulin sensitivity by sensitizing agents (metformin, TZD, among others) inhibits ATP production in the mitochondria.81 In any case, accumulated experimental evidence during the last decade of research have give us substantial support proposing that mitochondria seems to coordinate the development of metabolic complications linked to obesity, either promoting or inhibiting lipotoxic damage.
Certainly, it is necessary to conduct further integral research, which will determine whether or not mitochondrial dysfunction during obesity is a cause or the consequence of the metabolic imbalance which facilitates the development of T2DM in humans.
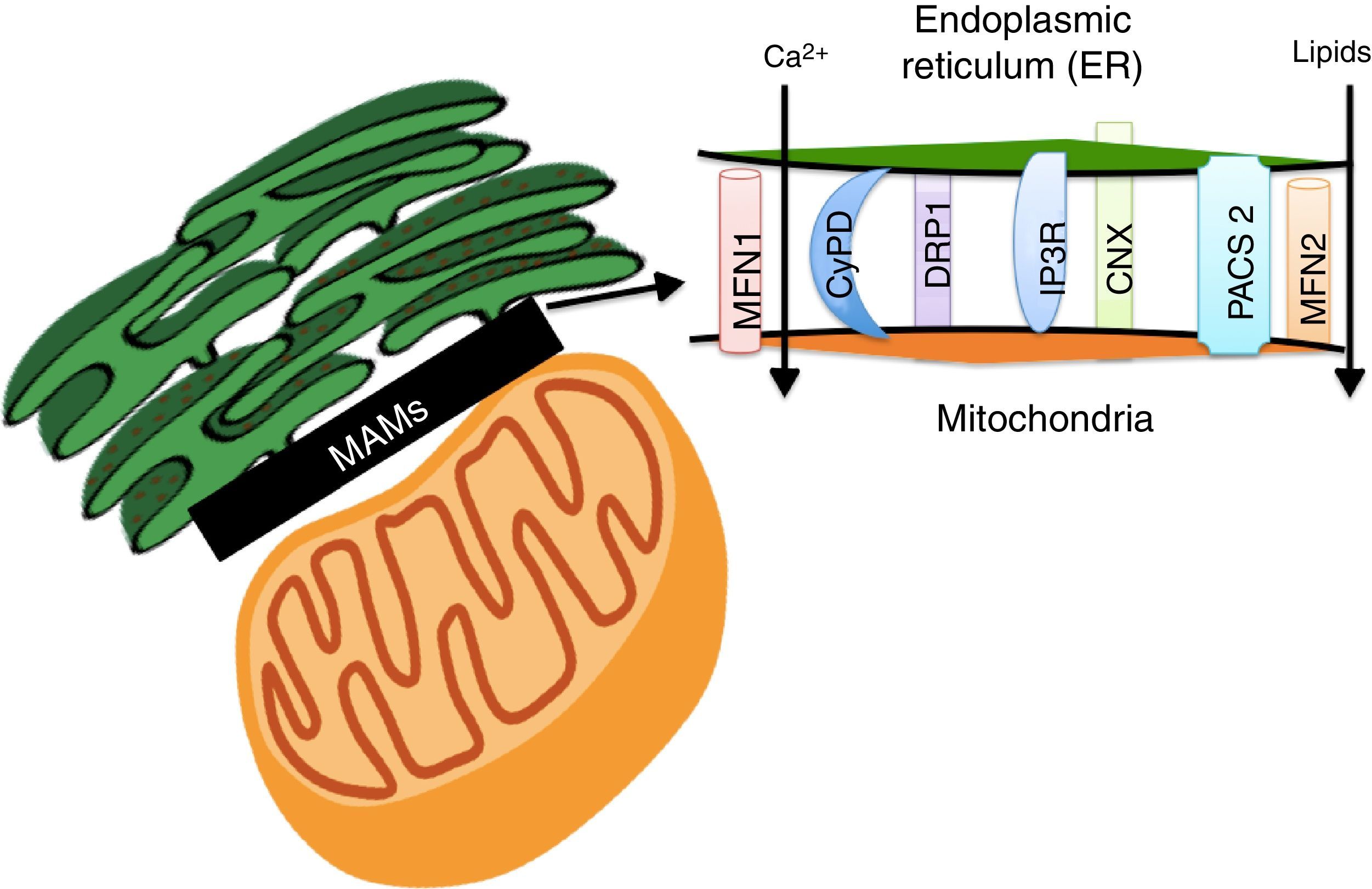
Lipids accumulation during obesity modulate MAMs formation and coordinate the development of diabetesER and the mitochondria interact metabolically and functionally through links between its membranes known as MAMs.82,83 Its function has been related to ER's phospholipids import to the mitochondria, the cellular response between the ER and mitochondria, and cell survival and death.17,84 MAMs constitute a microdomain which recruits different proteins of functional importance, among them: (1) enzymes which participate in the synthesis of lipids (acyltransferase cholesterol, diacylglycerol acyltransferase and phosphatidylserine synthase),84 (2) calcium-regulator proteins (inositol-1,4,5-tris phosphate receptor (IPR), ryanodine receptor (RyR), the voltage-dependent-anion-mitochondrial-channel-1protein (VDAC1) and the sigma-1receptor (SigR1)), (3) proteins which participate in protein folding (calnexin (CNX) and ERO 1L-α),84 (4) proteins involved in mitochondrial remodeling (dynamin-like 1 (DPR1) and mitofusins 1 and 2 (MFN 1 and 2)), and (5) the ER binding stabilizing proteins, PACS2 (Fig. 3).84
 are a microdomain made up of different proteins which are in charge of calcium homeostasis, transport of lipids, mitochondrial morphology and the oversight of cellular death. Their work has recently been associated with the coordination of energetic metabolism.")
The Mitochondria-Associated Membranes (MAMs) are a microdomain made up of different proteins which are in charge of calcium homeostasis, transport of lipids, mitochondrial morphology and the oversight of cellular death. Their work has recently been associated with the coordination of energetic metabolism.
The interaction between ER and the mitochondria has been recently proven to be regulated by the action of MFN 1 and 2 (Fig. 3).84 It has been proven that fibroblasts which do not express MFN2 display a reduction in the contacts between ER and the mitochondria, which recover when the MFN2 expression is reestablished in ER through genetic manipulation.85 Functional integration through MAMs allows calcium homeostasis regulation in the cell. In base conditions, Ca2+ concentration in the cell's cytosol (free 100nM Ca2+ versus 100mM in extracellular space) is maintained through active participation of the ER and the mitochondria. Cytosolic Ca2+ is captured to the ER by the action of the SERCA bomb and is taken by buffering molecules such as calsequestrin.86 Under stimulation conditions, Ca2+ is released from ER by the ryanodine receptor, activated by very low cytosolic Ca2+ concentrations, or through IP3R receptors (abundantly concentrated in MAMs)66 in response to phosphatidylinositol signals, produced when Ca+2 concentrations are low.86 Ca2+ released through IP3R flows to the mitochondrial matrix through mitochondrial calcium uniporter proteins (MCU),82 activating mitochondrial dehydrogenases activity which are a key element in ATP production.82,87 Nevertheless, an increase in ER's calcium flow to the mitochondria through IP3R promotes mitochondrial damage and apoptosis.87 In fact, MFN1, MFN2, phosphatidylinositol-3,4,5-triphosphate 3-phospatase (PTEN), promyelocytic leukemia (PML) and RE L-α oxydoreducin chaperone proteins (ERO-1 L-α) increase ER calcium release to the mitochondria, either phosphorylating or interacting with IP3R.88–92
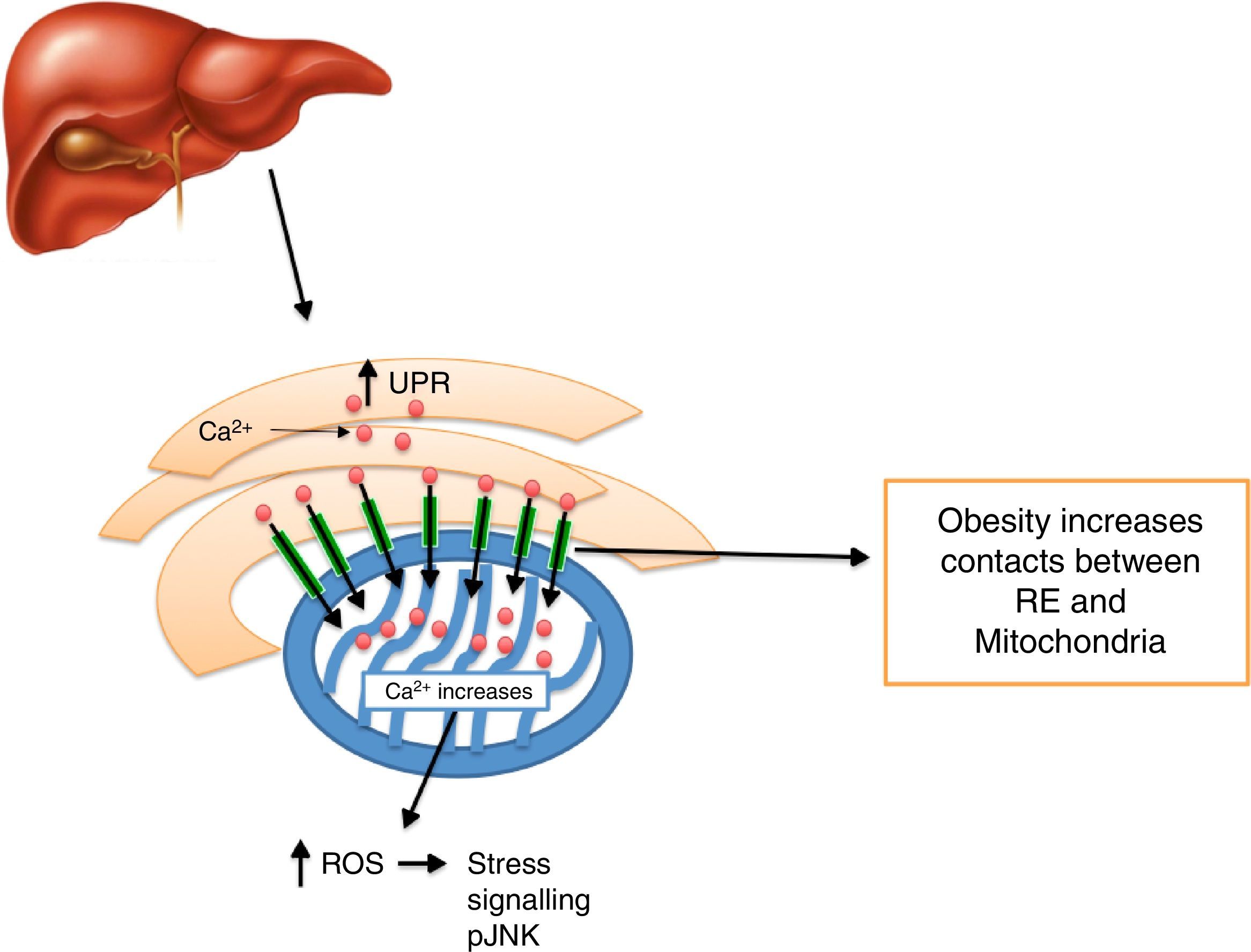
Calcium decrease at the ER level creates ER stress activation, including morphologic changes and an ER-mitochondrial bind increase,93 leading to an increase in mitochondrial activity.93 It has been noted that defects in MAMs microdomains’ proteins have been proven to promote ER stress previous to the development of mitochondrial dysfunction.41 This way, it is possible for defects in the conformation of MAMs to lead to alterations in calcium homeostasis or metabolic damage linked to ER stress, inducing a mitochondrial dysfunction. In this context, MFN2 deletion in rats and mice promotes insulin resistance and mitochondrial dysfunction linked to ER stress activation in skeletal muscle77,94 and liver95,96 and a decrease in MTF2 expression was proven in obese patients. It is worth stressing that obesity induced by high-fat dietary intake in mice who lacked MFN2 in anoerxigenic POMC neurons of the hypothalamus, created a decrease in ER and mitochondria contacts, resulting in ER stress, leptin resistance and a reduced energy expenditure.17 Contrarily, obesity induced by a high-fat diet intake in mice who lacked MFN1 and MFN2 in AgRP orexigenic neurons of the hypothalamus promoted a decrease in ER and mitochondria contacts, obesity, and increase in partial fat.18 Molecularly, it is suggested that an increase in ER and mitochondria contacts coordinated by MFN2 during obesity, increase Ca2+ flows from ER to the mitochondria through IP3R receptors, generating mitochondrial dysfunction, activation of the JNK pathway and insulin resistance19 (Fig. 4). All together, it seems that MAMs closely coordinate energy metabolism through their defects over calcium homeostasis, ER activation and mitochondrial function, molecular processes which are compromise during lipotoxicity induced by lipids accumulation during obesity.97
Strategies for obesity treatment and its effects in patients
Given the effect of obesity in the development of comorbidities, different strategies have been used with the purpose of inducing weight loss and its effect on the improvement of glycemic control in obese subjects with T2DM.98,99 However, keeping the weight off represents a greater challenge in obesity treatment.100 To this end, different interventions have been employed, such as diets, pharmacological treatments and surgery. As an example of the first intervention, in a meta-analysis study using data from the Cochrane Central Register of Controlled Trials and EMBASE from January 1981 to February 2013, anti-obesity drugs, food replacement and high-protein diets were proven to be linked to weight-loss maintenance compared to food supplements and exercise.100 On the other hand, pharmacological treatment has not been so successful, in part due to its limited success in the long term and lack of effect when the consumption is stopped, also the development of major side effects.101,102 One example in this scenario is the Fen-Phen (Fenfluramine/phentermine, serotonin/noradrenaline releasers), withdrawn from the market in 1997 because of its side effects, which included valve disease and pulmonary hypertension, and rimonabant (CB1 receptor antagonist), withdrawn from the market in 2008 because of its positive effect on the generation of severe depression and suicides.101 As a third intervention, there is bariatric surgery, which has proven to be effective in weight loss and even remitting T2DM or improving glucose levels in patients for over two years.103 We will now describe the effect of said intervention on the nervous system for the control of metabolism.
Bariatric surgery and its effect on the CNS for promoting weight lossIt is well-known that not all patients who lose weight in the obesity context are able to keep the weight off in the long term. For example, in the US, approximately 1 in every 6 subjects can maintain the loss of 10% weight in the long term, the rest suffer a relapse and continue to be at risk of T2DM generation.104 Since its beginnings in the 50s, bariatric surgery, including Roux-en-Y gastric bypass (RYGB), adjustable gastric band (AGB) and biliopancreatic diversion have been the most effective interventions for the treatment of obesity in a population with morbid obesity (BMI ≥40 or ≥35).105,106 These surgeries allow weight loss through caloric restriction and/or malabsorption of calories and reverts or reduces comorbidities like T2DM, hypertension and hyperlipidemia, among others.105,107
Interestingly enough, the effect of bariatric surgery intervention has over the nervous system's ability to regulate the metabolism of the body has recently been demonstrated. Studies have shown high activity, specifically in the hypothalamus region of the brain of women who have undergone RYGB surgery when they are shown images of hyper-caloric food, regarding non-obese women, which correlate with a low score in hunger and a high score in satiety compared to those with normal and obese.108 Molecularly, the melanocortin 4 receptor (MC4R) has been proven to be a target responsible for weight maintenance in individuals who are heterozygous for this gene.109
Also, even though bariatric surgery allows for the reduction of body weight, its effect in the long run depends in large part on obese subjects. The reason for this effect may be linked to a change in hormone liberation from the intestine and stomach, which regulates the body metabolism. RYGB and VSG bariatric surgeries are known to increase the release of pepri YY (PYY), released by L cells from the distal small intestine,110,111 but it does not have an effect during caloric restriction or due to a gastric band surgery.106,112,113 Weight loss is greater in patients who underwent RYGB surgery and who present an increase in PYY.114,115 PYY has an anorexigenic effect, acting at a solitary tract nucleus and ARC level116 to promote an increase in energy expenditure and delay stomach emptying.117 Another peptide secreted by L cells is GLP-1 (glucagon-like peptide-1), which inhibits glucagon release and slows gastric emptying, also reducing food intake by stimulating the hypothalamus and brain stem hormones, among other regions of the brain like striatum and black matter.106,118 GLP-1 has shown similar responses to PYY after RYGB and VSG surgeries.106,119A third hormone which suffers changes after gastric surgery is ghrelin, which is released from the stomach and inhibits food intake, inducing the activation of the hypothalamus. Ghrelin is reduced after VSG surgery,120 increases after gastric band surgery and remains without changes, increases or decreases, after RYGB.106,121,122
Finally, basic science experiments have shown non-conclusive target results additional to the effect of the bariatric surgery over the nervous system function. Among them, reports suggest that there are not any changes in the neuropeptide expression which promotes food intake, including AgRP and NPY,123 while other reports show a decrease in NPY expression,124 or an NPY and AgRP increase.125 All together, more evidence is required to conclusively determine the effect of bariatric surgery over body weight regulation through the anorexigenic, orexigenic neuropeptides and GLP-1 function expressions in the CNS.
Possible strategies for MAMs remodeling as obesity treatmentOne of the most outstanding pharmacological strategies to control lipotoxic damage induced during obesity is the use of chemical chaperones. Chemical chaperones are agents which avoid ER stress and improve mitochondrial function. These chaperones produce changes in MAMs proteins, lowering of the binds between ER and the mitochondria, the increase of oxidative stress and Ca2+ flow to the mitochondria.19 In mice models, chemical chaperones including 4-phenyl butyric acid (4-PBA), a low-molecular-weight molecule, and taurine ursodeoxycholic conjugate acid (TUDCA), an endogenous biliary salts derivate,16 and R, S-O-(3-piperidino-2-hydroxy-1-propil)-dihydrochloride of amidoxime nicotinic acid (BGP-15), have shown to be effective in protecting against the ER stress activation favoring insulin sensitivity. In this context, results in cellular culture as well as in genetic animal models of obesity (mice ob/ob), have show that chaperones reduce ER stress, normalize glucose levels and increase insulin sensitivity.16 In particular, BGP-15 is the most promising candidate to improve insulin sensitivity, since it is comparable to other insulin sensitizers tested, like rosiglitazone,126 in addition to having effects in patients with diabetes.127 Other new drugs which module ER stress response are being tested in animal models, such as celastrol, which is a molecule extracted from the Tripterygium wilfordii plant that reduces ER stress and acts as a leptin sensitizer, and has great potential as anti-obesity therapeutic agent.128
Even though it has not been proven in humans, evidence suggests that protein modulation, which comprises the MAMs microdomain, may be a therapeutic target to improve metabolism and reduce oxidative stress and the inflammatory response observed in obesity.129
ConclusionsPOMC and AgRP neurons of the ARC nucleus of hypothalamus represent a functional node which regulates corporal energy homeostasis and is the target of pathological alterations during obesity which predispose to the development of diabetes. One of the most significant alterations caused by obesity is the increase of lipids, which generates changes in MAMs proteins recruitment and spatial conformation, promoting ER stress and an increase in calcium flow between ER and the mitochondria. ER stress activation is known to be linked to ROS increase and inflammation, which in turn promotes the development of insulin resistance, glucose intolerance and diabetes. A strategy to revert these alterations is through bariatric surgery, since it promotes changes in hormones released in the intestine like PYY, ghrelin and GLP-1. Another strategy which may be available in the future for obese patients is chemical chaperones, which lower ER stress and improve insulin and leptin sensitivity. We suggest that changes or alterations in MAMs remodeling represent a pathological mechanism in the context of obesity, where molecular systems implicated in ER stress activation and mitochondrial function contribute to the control of metabolic homeostasis. Thus, thoroughly addressing the characterization of MAMs microdomain may enable us to find new therapeutic targets for obesity treatment.
Conflict of interestThe authors have no conflicts of interest to declare.
FundingCONACYT Ciencia Básica No. 168236.