


Introduction
Lipids and their metabolic derivatives constitute an important part in living cells structure and physiology, but until recently were not sufficiently studied. In a relatively short period, a growing field of technologies has provided an immense amount of data regarding the role of fatty acids as well as and many other molecular species within the living cell, thus giving rise to a systems level field of study: lipidomics. The relevance of this body of knowledge is paramount in medicine, since it already became clear that a wide variety of diseases including Mycobacterium infections and other infectious diseases, Diabetes, Alzheimer´s and cancer hold particular imbalances in lipid metabolism. The main families of compounds within this scope are: fatty acids, glycerolipids, glycerophospholipids, sphingolipids and sterols. In this review we will focus on the emerging knowledge of the role of lipids in infectious disease.
Lipid research in a traditionally water-based biomedical research environment was difficult because of a lack of appropriate research tools. However, technological innovations, especially in mass spectrometry, as well as an increased interest in lipid metabolism because of lipid-centered chronic diseases, including the metabolic syndrome, boosted this research area, recently coined as lipidomics. The lipid profile of a sample, whether it concerns a liquid sample, a cell or tissue, is known as the lipidome. Lipidomics goes beyond, as it includes the interactions between lipids, proteins and other metabolites.1 Current technology allows in situ lipid profiling from tissue sections with specialized mass spectrometry techniques,2 and recently developed Coherent Anti-Stokes Raman scattering (CARS) microscopy enables the study of lipid dynamics in living cells.3
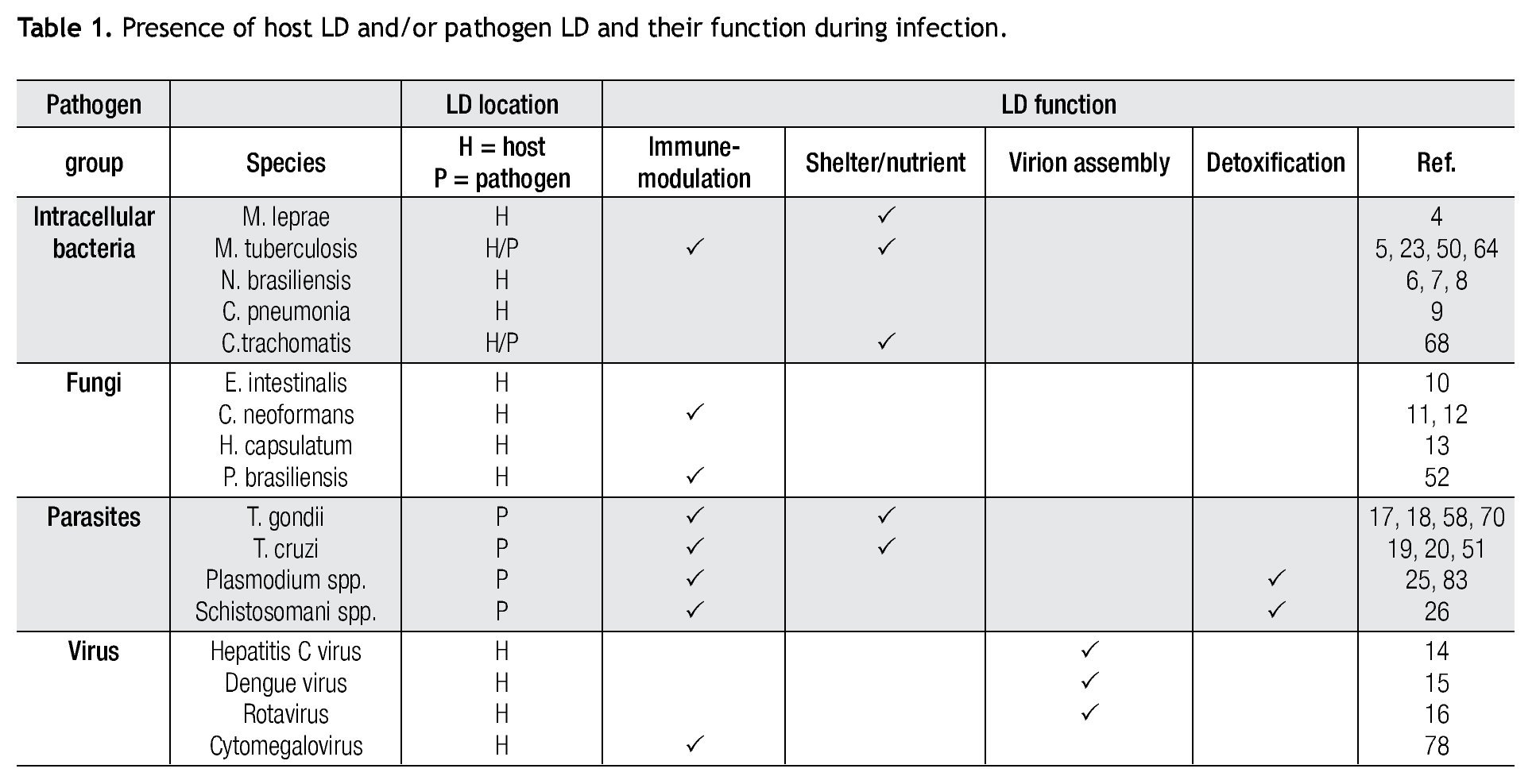
The burgeoning field of lipidomics also impacted the investigation of the immunology of infectious. Renewed attention is being paid to the appearance of intracellular lipid droplets (LDs) within host cells infected by the intracellular forms of certain pathogens, a phenomenon that has been observed in all of the four main groups of pathogens (viruses, bacteria, fungi and parasites) (Table 1). In most cases, it seems that the pathogen takes advantage of the formed LDs. The German physician and pathologist Rufdolf virchow (1821 - 1902) described lipid accumulating cells in biopsies from lepromatous leprosy lesions, known as Lepra cells or foamy cells that are distinctive for this severe form of leprosy.4 The causative pathogen Mycobacterium leprae can be found within these foamy cells. Another mycobacterium species, M. tuberculosis, that asymptomatically infects almost a third of the world´s population, causes a lipid pneumonia when it reactivates after a latent period as a secondary tuberculosis.5 Also various Nocardia spp. induce foamy cells, which typically are located just below the fibrosis ring that delimits the multilocular microabscesses (Figure 1).6-8Chlamydia pneumoniae is another intracellular bacterium that induces foamy macrophages, which have been associated with the induction of atherosclerosis.9 Certain fungi such as, Encephalitozoon intestinalis,10Cryptococcus neoformans,11,12 and Histoplasma capsulatum13 cause an abundant accumulation of LDs within leukocytes early in infection, which seems related with the production of lipid immunomodulators. In the case of viral infections produced by hepatitis C (HCV), dengue virus and rotavirus, LDs are important for the assembly of infectious virions.14-16 Regarding parasites: Toxoplasma gondii17,18 and Trypanosoma cruzi19,20 have been observed clustering around LDs within the host cell, while Plasmodium berghei induces LDs within hepatocytes.21 Nippostrongylus brasiliensis is a helminth that induces foamy cells transiently early in infection.22 Some pathogens also accumulate LDs within themselves during the infection, for example M. tuberculosis,23Candida parapsilosis,24 and various blood-feeding parasites (Table 1).25,26 Despite the findings listed above, there is still an overall gap in knowledge about the importance of LDs in certain infections. Therefore, the aim of this review article is to call attention to this area. First, we present the new concepts on general LD composition, function and biogenesis generated by recent lipidomics data. Secondly, we describe various mechanisms reported to explain the presence and function of LDs in various infections.
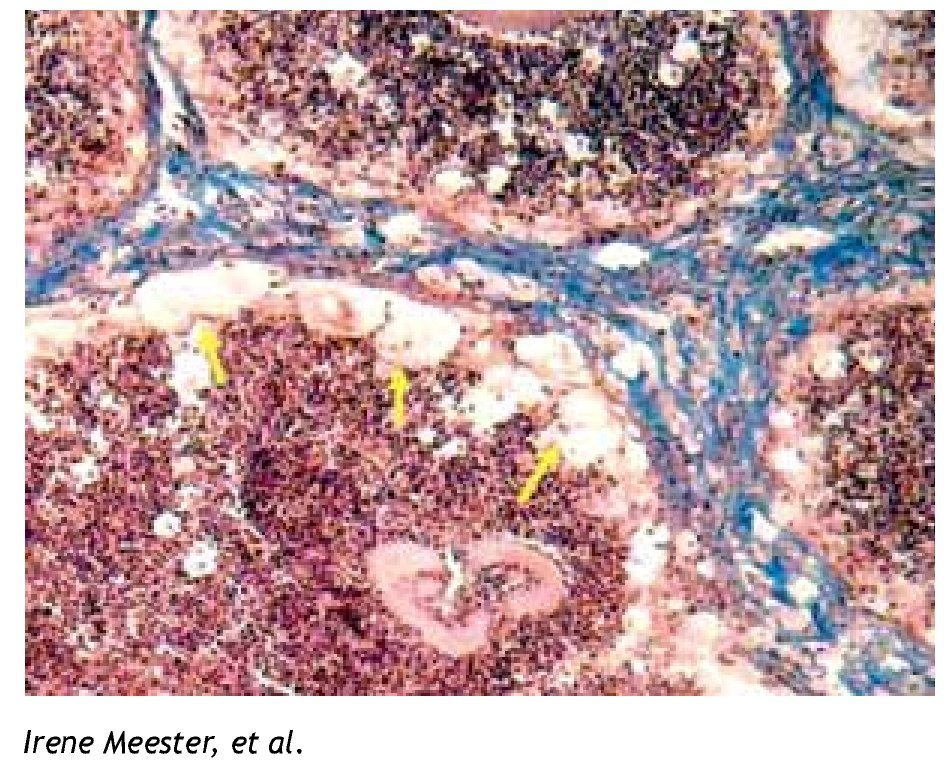
Figure 1. Foamy cells (arrows) in a 90-day actinomycetoma of the mouse foot pad induced by Nocardia brasiliensis.
Basics of LD
Composition and function
The traditional idea that LDs are simple, inert lipid accumulations has been replaced by a new concept that recognizes LDs as more complex, dynamic organelles which are heterogeneous, not only between cells but even within the same cell.27 Previous reviews have described in detail this new information on LDs.28-31 Here we summarize the new perception of a LD, highlighting major departures from the traditional view and discussing the impact for its functionality.
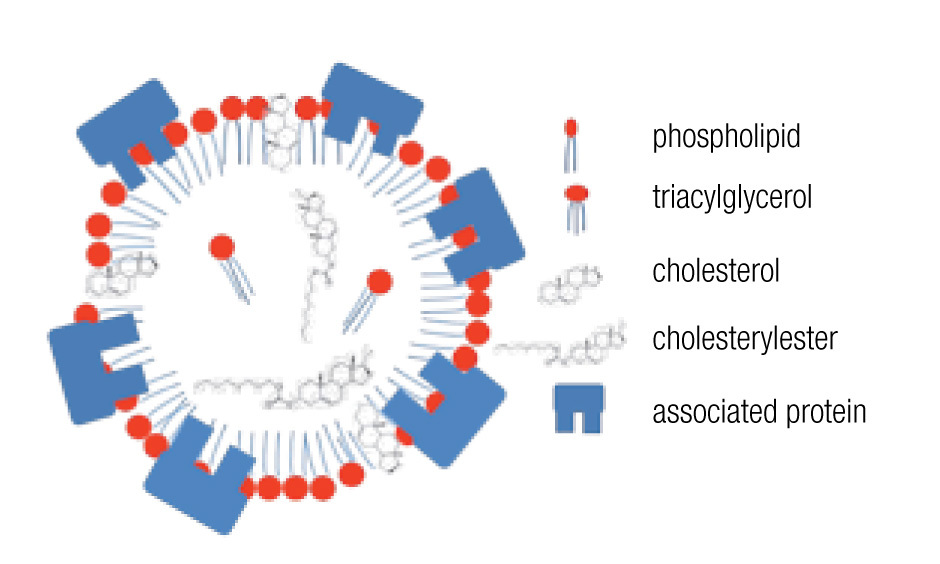
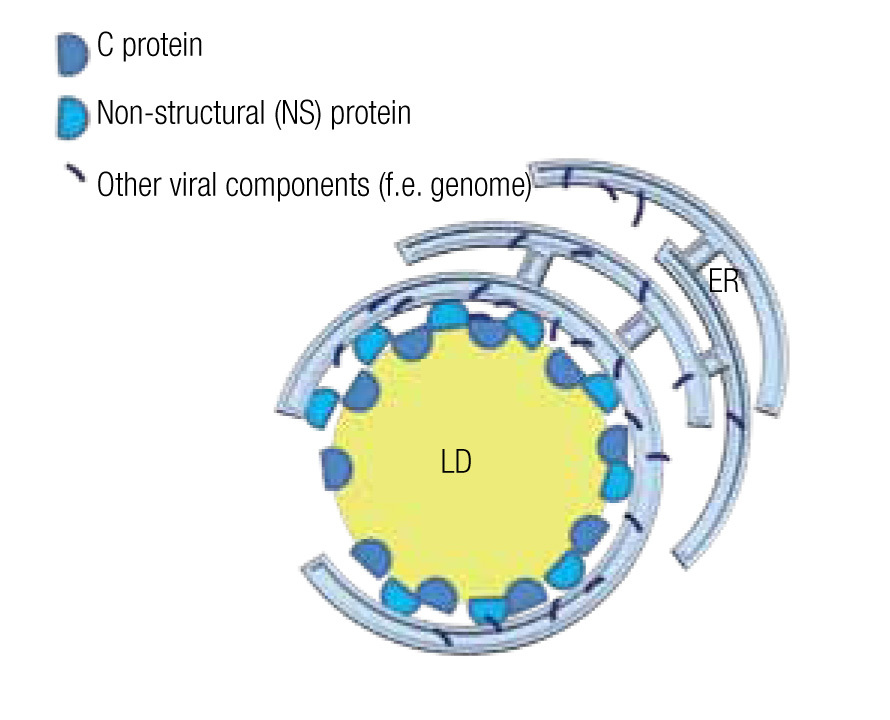
LDs are composed of a core of neutral lipids (mainly triglycerides and cholesterol esters), which are delimited by a phospholipid monolayer and associated proteins, resulting in a structure similar to lipoproteins (Figure 2).28 Depending on the function of the LD, different neutral lipids are present in the LD interior and different proteins are associated with the LD.30 Likewise, the size of the LD varies notably: from the large unilocular LD present in white adipose tissue adipocytes that serve for long-term storage to the small multilocular LDs that facilitate access to and mobilization of their content. LD size is dynamic, amongst other through processes of fusion and fission. Size changes have important consequences for the ratio between neutral lipids and phospholipids. Upon LD fission, an increase in surface area occurs and the LD instantly requires more amphipathic lipids; whereas a sudden excess of these lipids results after an LD fusion process. How these dilemmas are resolved remains poorly understood, but it is clear that LD size is well-regulated.29,32 One known size-regulatory protein is seipin.33 Mutated seipin leads to congenital generalized lipodys-trophy, characterized by a lack of normal adipose tissue, but accumulation of fat in liver and muscle.34
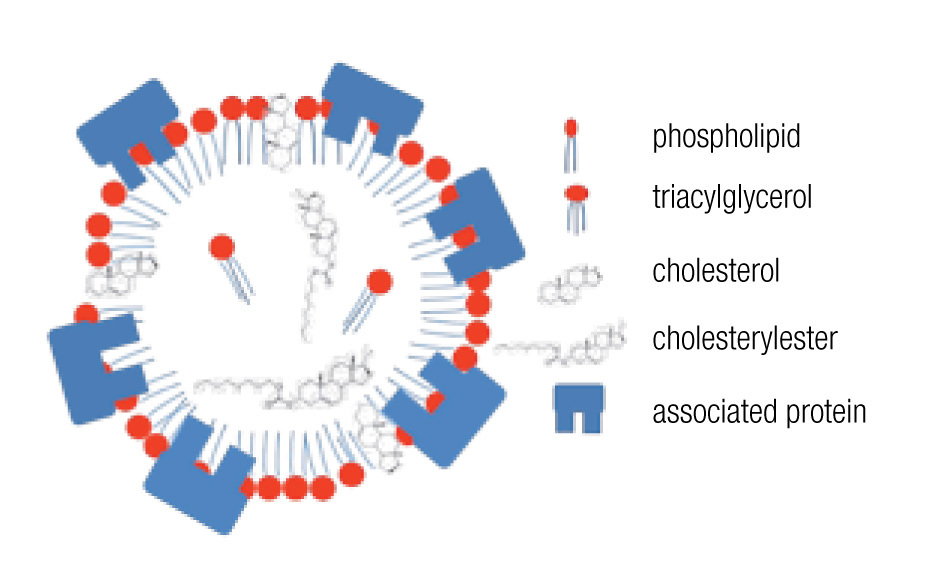
Figure 2. General lipid droplet structure; based on Guo.28
Proteins of the PAT/PLIN family are consistently found on LDs, and therefore are often used as LD markers. PAT/ PLIN family proteins include PERILIPIN (PLIN1), adipo-philin/adipose differentiation-related protein (ADRP/PLIN2), and tail-interacting protein (TIP47/PLIN3), as well S3-12/PLIN4 and oxidate-tissue-enriched PAT protein (OX- PAT/PLIN5).35 PLIN proteins are in charge of stabilizing LDs and controlling the mobilization of their content.31,36 Depending on the metabolic phase of the LD, enzymes for lipid synthesis or degradation may be associated with an LD, facilitating either lipid storage or mobilization.29,37 Vesicle trafficking proteins, such as Rabs, soluble N-ethylmaleimide sensitive factor attachment protein receptors (SNAREs) and motor proteins, are often found on LDs and probably involved in intracellular lipid transport, fusion mechanisms, and interactions with other organelles.27 This is consistent with the fact that LDs interact with other subcellular components such as peroxisomes and mitochondria,30 endoplasmic reticulum (ER),37,38 and the cytoskeleton.37,39 Surprisingly, non-folded proteins, histones, and cytokines have been found at LDs, which suggests that LDs could serve as a safe storage site for dangerous or temporarily unnecessary proteins.40,41 in leukocytes, even proteins involved in the translation process from mRNA to proteins have been found within LDs.37,42 This phenomenon was considered an experimental artifact until ultramicroscopy data confirmed the presence of ribosomes and ER within these leukocyte LDs, which generates highly efficient lipid producing compartments.37
Biogenesis of lipid droplets
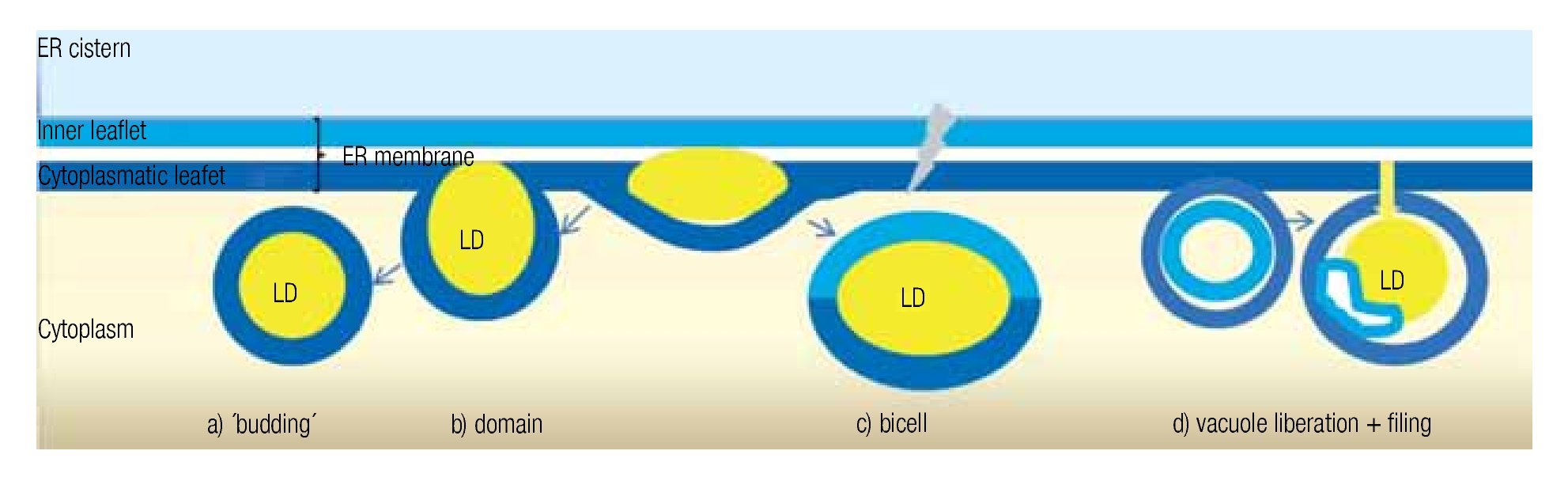
LD may occur in all cells, both prokaryotic and eukaryotic. Their formation is normally membrane-dependent: in prokaryotes, they are generated at the cell membrane,43 while in eukaryotes the ER is the site of LD biogenesis. Four main models have been proposed to describe LD biogenesis in eukaryotes28 (Figure 3); 1) in the widely accepted ´budding´ model, neutral lipids accumulate between the two layers of the lipid bilayer and when the ´bud´ is released it remains surrounded by the cytoplasmic layer of the ER membrane; 2) The domain model differs from the previous one in that the ´bud´ is never released but remains attached to the ER. This model could facilitate size change of LDs: during LD growth, it could provide the extra neutral lipids and "absorb" the excess of amphipathic lipids, and vice versa. 3) in thébi-cell´ model, the lipids accumulated within the bilayer are excised at the extremes. This is the only model in which the cisternic layer of the ER membrane participates in delimiting the LD.44 4) The vesicular model proposes that first an ´empty´ vesicle is liberated from the ER. Subsequently, the intramembranal space is filled with neutral lipids, pushing the cisternic layer aside. As such, this is the only model that should have remains of the cisternic layer within the LD. So far, there is no model that clearly explains how ER ends up within the LD.36
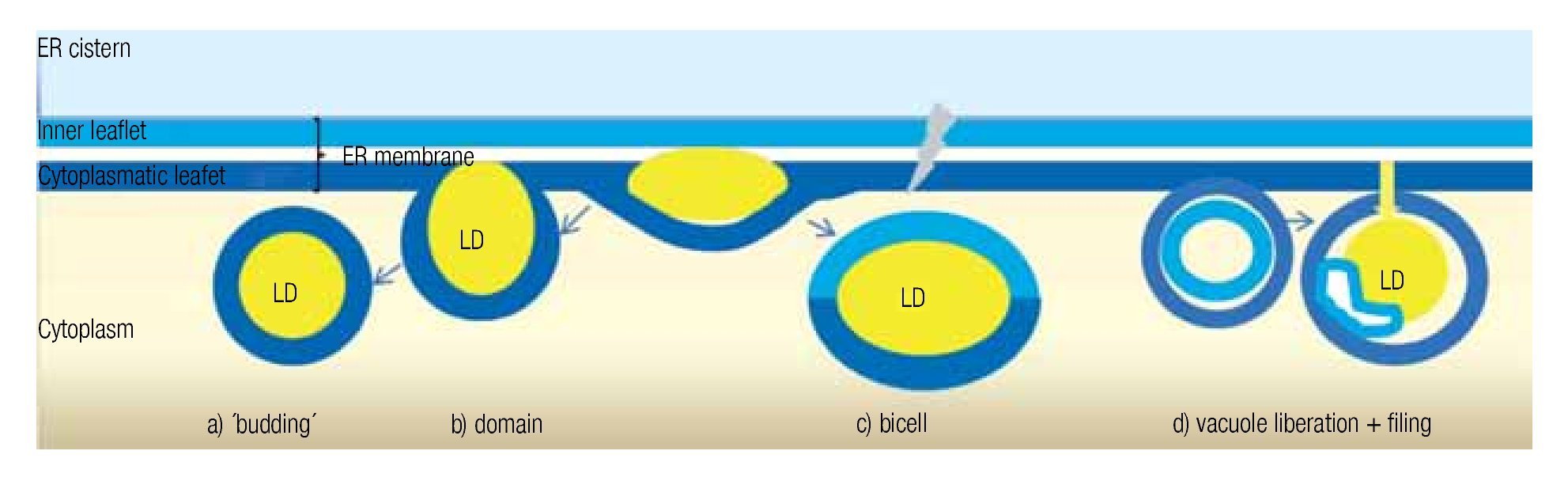
Figure 3. Theories of lipid droplet biogenesis.
Host LDs facilitate the production of lipid immunomodulators
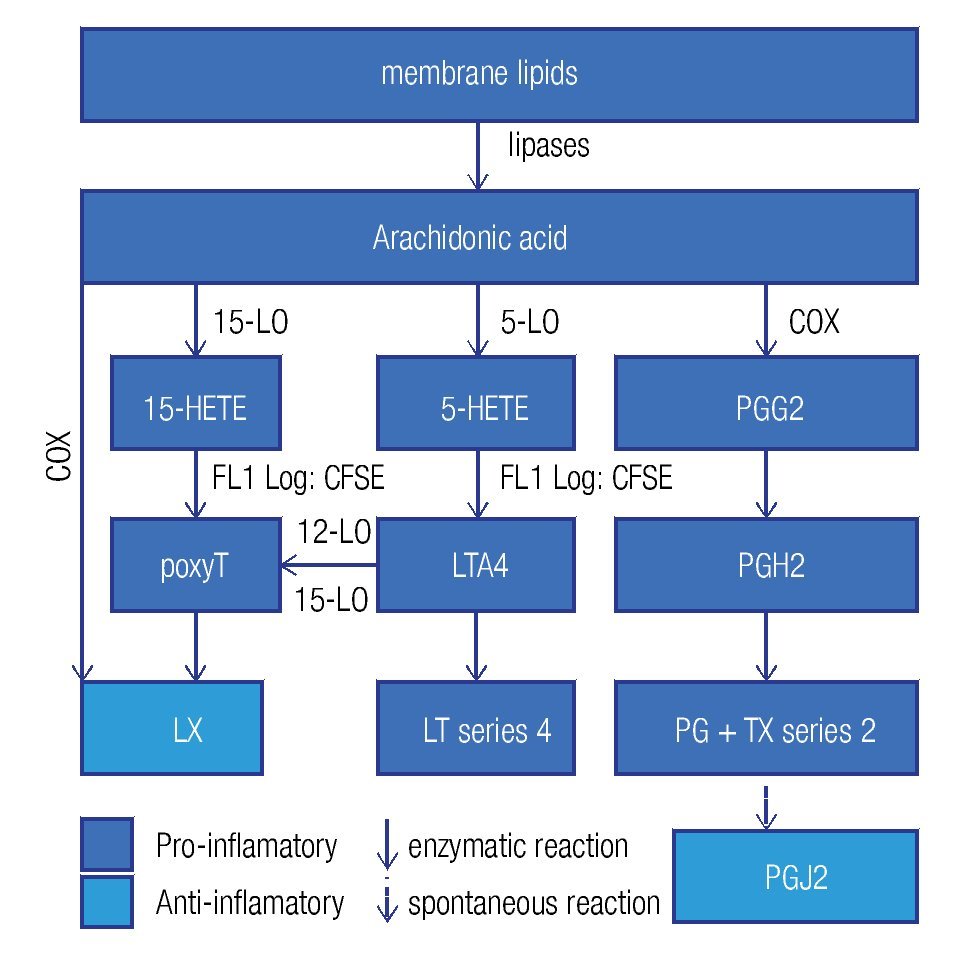
Enzymes that generate eicosanoids, such as prostaglandin H2 synthase/cyclooxigenase (PGH2 synthase/COX)45 and lipoxygenases (LO)46 have been localized at LDs. Generally, eicosanoids are involved in the classical signs of inflammation: redness (due to vasodilatation), swelling (a consequence of an increased vascular permeability), pain (PGE2 stimulates pain neurons), heat (PGE2 is pyre-tic) and finally loss of function. They are classified in a) prostanoids (which include prostaglandines [PG], pros-tacyclins [PGi] and thromboxanes [TX]), b) leukotrienes (LT), and c) lipoxines (LX). Eicosanoids originate from arachidonic acid and function as immunomodulators. Figure 4 summarizes the synthesis pathways of the different types of eicosanoids. Eicosanoid synthesis starts with the liberation of arachidonic acid from membranes by phospholipase C or A2. Next, PGH2 synthase/COX catalyzes the conversion of arachidonic acid into PGH247 in a two-step way: cyclooxygenation and peroxidation. The former reaction, cyclooxygenation, is responsible for the best known synonym for this enzyme: COX. Two isoforms of PGH2 synthase are recognized: COX-1, which is constitutively expressed in many cells at low levels, and COX-2, or inducible COX, which is mostly associated with inflammation, as the cells, which express COX-2, subsequently convert PGH2 in pro-inflammatory prostanoids. However, when expressed at high levels, eicosanoids may also be anti-inflammatory or modulate the adaptive immune response.
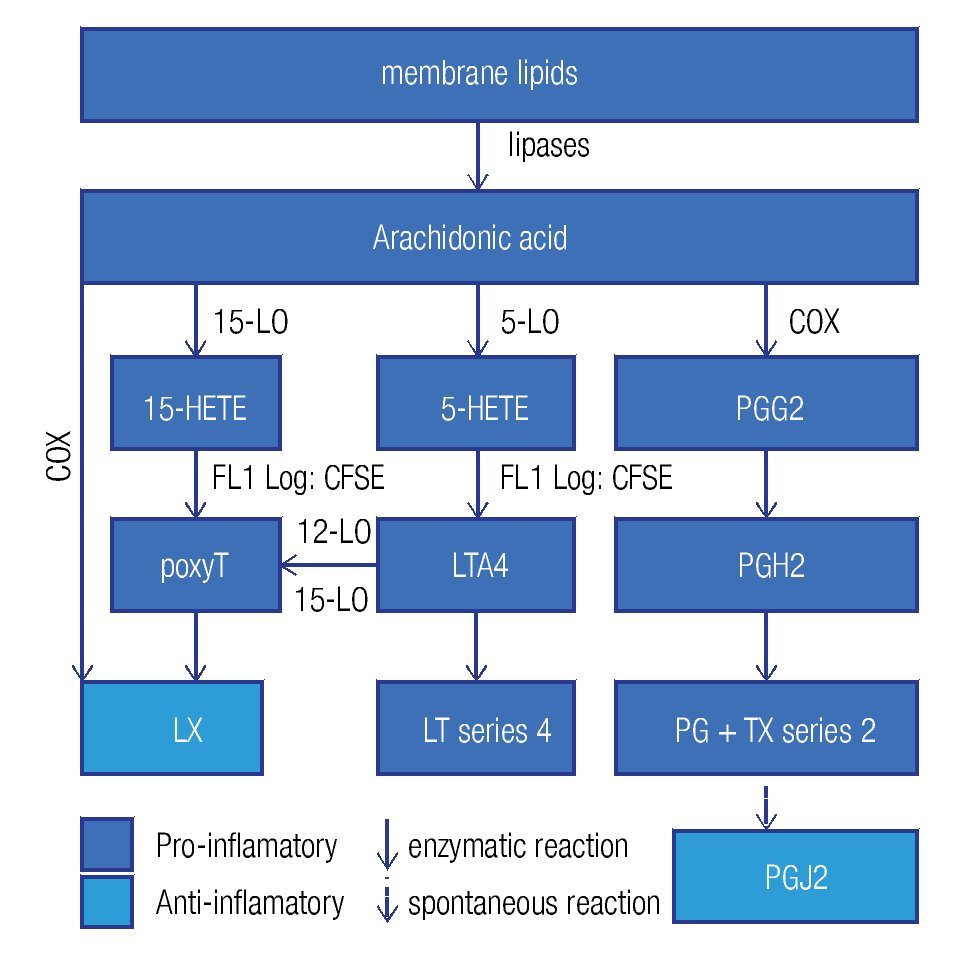
Figure 4. General lipid droplet structure; based on Guo.28
Pathogens taking advantage of eicosanoid immunomodulators
High levels of PGE2 may generate derivatives like PGD2 and PGJ2, which are considered anti-inflammatory and pro-resolution.48 PGJ2 is an endogenous ligand for the nuclear receptor peroxisome proliferation-activated receptor gamma (PPARg), which is not only a key regulator of lipid metabolism but also has anti-inflammatory effects.49 Therefore, COX-2 presents dual activity: pro-inflammatory in the early phase of inflammation and pro-resolution in later phases. Furthermore, eicosanoids have been shown to modify the adaptive immune response, where high PGE2 levels may divert the Th1 response to a Th2 reponse.50 It seems that some pathogens take advantage of the immunomodulation by eicosanoids. In various infections, including the ones induced by parasites (Trypanosoma cruzi),51 fungi,11,52 and intracellular bacteria,53,54 high levels of PGE2 have been found. Excessive PGE2 levels were related with immunosuppressive effects11 and a switch of the immune response to Th250 which is less effective against intracellular pathogens. Some species of fungi, such as Candida albicans, Cryptococcus neoformans11 y Paracoccidioides brasiliensis,52 produce PGE2 themselves via transcellular biosynthesis. Transcellular biosynthesis is a mechanism in which a biosynthesis pathway is carried out partly in one cell generating an intermediate metabolite, which is transferred to another cell that finalizes the route and genera- tes the end product. The fungi take up arachidonic acid from their environment, the host cell, to produce PGE2, which seems a mechanism of immune evasion and important for their survival. Therefore, in some infections, COX inhibitors may help in recovering from infections.11,51
Recently it has become clear that the LX55,56 and the eicosanoids derived from w-3 polyunsaturated fatty acids, for example eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA)57 are anti-inflammatory and actively stimulate resolution. LX inhibits the pro-inflammatory response during chronic inflammation.58 LXA4 reduces pain and vascular permeability, and diminishes the recruitment of neutrophils, while it augments the recruitment of monocytes and the activation of macrophages, but not their pro-inflammatory activities.59
The parasite Toxoplasma gondii contains a protein with the enzymatic activity of 15-LO, enabling the conversion of host-derived arachidonic acid into the intermediate 15-HETE, which is subsequently transferred back to the host cell to be converted into a LX through a mechanism of trans-cellular biosynthesis.60 in addition to the anti-inflammatory effects of LXs,58,61 they also diminish iL-12 production and consequently the cellular immune response.62
Tuberculosis offers an intriguing example of the different effects of eicosanoids. The infection by M. tuberculosis causes microdisruptions in the cell membranes, which would normally lead to necrosis, but PGE2, abundantly present in the foam cell,53 promotes membrane repair to protect against necrosis. On the other hand, PGE2 induces apoptosis and consequently antigen cross presentation by antigen presenting cells to T cells thus stimulating adaptive immunity. On the other hand, virulent strains induce the expression of LXA4, which inhibits PGE2, allowing necrosis and thus the liberation of pathogens, which can infect other cells and disseminate the infection.63,64
Host LD as shelters and/or nutrition sources for pathogens
The lipids present in LDs in infections are not undigested remains of the pathogens, but seem to originate from the host. This has been demonstrated in tuberculosis. Peyron and collaborators stained foamy cells induced by M. tuberculosis with Nile Red, a fluorochrome that binds to lipids, but its emission spectrum varies according to the type of lipid to which it is bound. This way the presence of mycobacterial lipids within the LD could be discarded.23 Likewise, the caseation in tuberculosis lesions is primarily composed of host-derived neutral lipids and not of myco-bacterial cell wall lipids.65 The LD might form a shelter rich in nutrients and energy. Electron microscopy studies demonstrated that M. tuberculosis in infected foam cells translocates to the LD and, once inside the LD, accumulates its own lipid inclusions, enters a non-replicative phase,23,66 and changes to a lipid based metabolism as reflected by the increased expression of isocitrate lyase.67
Chlamydia trachomatis, a pathogen that causes ocular and genital infections is another bacterium that depends on host lipids, especially to form the membranes of its replicative form, the reticular body. The reticular bodies multiply within an endosome known as the parasitophorous vacuole. The large amounts of phospholipids that are required for the microbial replication are obtained in part from host exocytosis vesicles rerouted to the parasitophorous vacuoles68 and in part from internalized LDs. For the latter, the bacterium secretes the protein Lda3, which recruits LDs and assists in their translocation into the parasitophorous vacuoles. Once internalized, LDs are consumed for membrane synthesis.68
Toxoplasma gondii is an obligate intracellular parasite, which depends on the host for its cholesterol needs.69 Host cholesterol, derived from low-density lipoproteins or free cholesterol is translocated to the parasitophorous vacuole and subsequently to the parasite. The intercepted cholesterol and other lipids are selected and modified in order to generate the particular lipids of the parasite.70 Cholesterol is incorporated in parasite membranes and into the apical organelle involved in invasion, known as the roptri.71T. gondii esterifies excess cholesterol to be stored in intraparasitic LDs.71,72
Host LD are assembly sites for viral particles
The viral genome encodes non-structural (NS) proteins and structural proteins. NS proteins have a function during the replication cycle within the host cell, while structural proteins form part of the viral particle. However, the structural core protein, or protein C of several virus of the family of Flaviviridae, such as the hepatitis C virus (HCV) and dengue virus, not only forms the capsid that wraps the genome, but also has a function during the intracellular phase. Notably, it associates with host LD by means of amphiphilic a-helixes.14,15 Likewise an NS protein, the NS5 protein in the case of HCV, although mainly localized in the ER, also interacts with LD by such amphiphilic a-helixes.73 Mutations in the amphiphilic domains of these viral proteins not only disable the interaction with the LD but also the assembly of viral particles.14 The importance of LD in the assembly of viral particles is further demonstrated by the fact that drugs that inhibit fatty acid synthesis or LD biogenesis cause a drop of greater than 10 to 1,000 fold in the formation of infectious viral particle in vitro studies.15,16,74 Several examples of these inhibiting drugs include TOFA (an inhibitor of acetylCoA carboxylase, the first step in fatty acid synthesis), C75 (a fatty acid synthase inhibitor), triacsin C (an inhibitor of long fatty acyl CoA synthetase), or iBMX (a phosphodiesterase inhibitor that diminishes leukotriene synthesis).
In the case of HCV, a model has been proposed to explain the roles of the mentioned proteins and the LD in the assembly of viral particles. The NS5 protein, localized principally at the ER similar to most viral components, associates with the LD where the capsid protein is stored and assembly centers are generated (Figure 5).75,76 Mutations in the NS5 protein yield higher density viral particles of less infectious capacity.75,77 LDs are also important for the release of these viral particles and entry into the next host cell. In fact, HCV particles are continuously associated with lipid structures. Less HCV particles are released when one interferes with the formation of lipoproteins, whe- ther by inhibiting the expression of apolipoproteins A, B or E or by inhibiting the lipid transport across ER membranes.14,77 Once in the blood stream, most HCV particles are bound to low-density lipoproteins, which augments their infectious capacity75 as it facilitates the entry into the next host cell, a process that can be inhibited by antibodies directed against apolipoproteins A and E.14
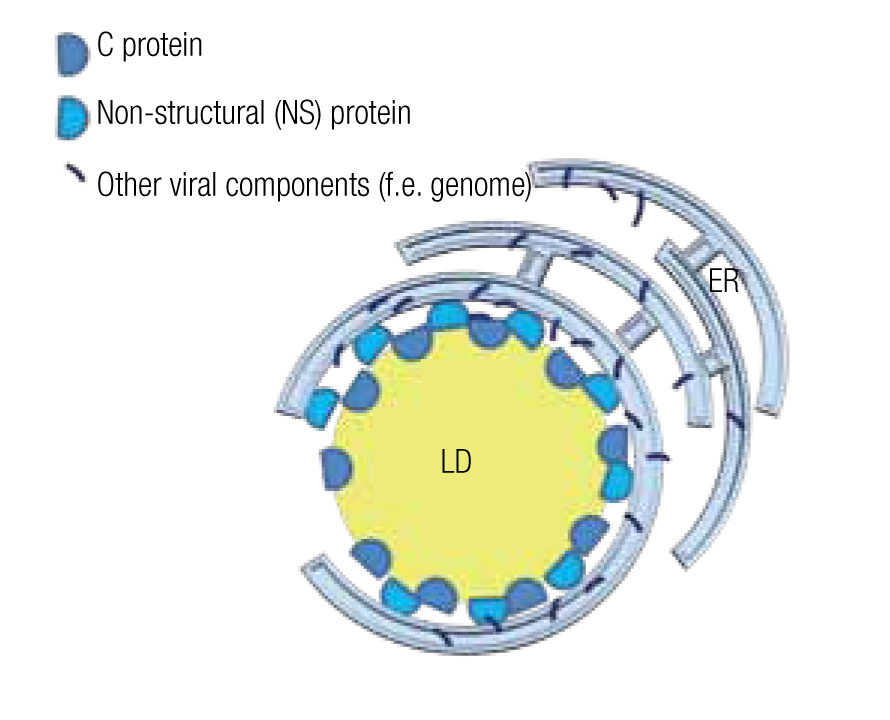
Figure 5. Model of a lipid droplet as a viral assembly center; based on Ogawa75 and Fukasawa.76
The importance of LD or lipid metabolites for the production of viral particles has been reported for evolutionary divergent viruses such as HCV,14,76 dengue virus,15,78 rotavirus,16 cytomegalovirus (CMV)79 and influenza virus.74 Research suggests that the antiviral defense system has developed a mechanism to interfere with the interaction between viral components and LDs. Type i interferons induce the antiviral protein, viperin, that localizes to ER and LD;80 its antiviral mechanism remains to be elucidated but it seems strategically located to interfere with the assembly of viral particles.
Pathogen LD: detoxifying agent for the pathogen and immunotoxic to the host
various blood-feeding parasites take advantage of the abundantly present hemoglobin as a protein source. However they have to deal with the liberated lipophilic free heme which is toxic by inhibiting certain enzymes, destabilizing membranes and generating lipid peroxidation products.81 Different organisms have evolved different mechanisms to protect against the nocive effects. For example, human beings detoxify heme by rapidly transferring free heme to the microsomal compartment where heme oxygenase releases iron and the fragmented porphyrin ring becomes bilirubin. Another mechanism, applied by various blood-feeding insects, helminths and protozoa, including Schistosoma and Plasmodium spp., is the crystallization of heme into hemozoin. This is also known as malaria pigment, as this brown pigment is found in Plasmodium infections.82 This detoxifying strategy starts with the conversion of heme into ferriprotoporphyrin-iX dimers, which is further crystallized in a process in which histidine-rich proteins and especially LDs play important roles.82, 83
Although hemozoin was long considered biologically ´inert´ and mainly used as a diagnostic tool, recent data indicate that it inhibits effector functions of monocytes and macrophages that have phagocyted parasite-infected erythrocytes or shed hemozoin from ruptured erythrocytes.84 Hemozoin inhibits the expression of integrins, the differentiation toward antigen-presenting cells, and killing ability,85 but stimulates a continuous release of pro-inflammatory cytokines.86 it has been suggested that heme-catalyzed lipid peroxidation products are responsible for most of the immunotoxic effects.87 Delipidized hemozoin has lost its immunotoxic potential, emphasizing the importance of lipids in relation to immunotoxic effects of hemozoin.88
How do pathogens induce LD?
In the previous sections, we presented the importance of LDs in infections, yet relatively little is known about how pathogens induce LDs. in the section on how pathogens take advantage of eicosanoid immunomodulators, we explained how T. gondii promotes the production of lipoxins via a transcellular biosynthesis pathway. Several additional cases have been shown in which the LD induction process received attention.
For tuberculosis, the cell wall component trehalose-6,6´-dimycolate, coated onto inert beads, was shown to induce foamy macrophages.89 Additional mycobacterial cell wall components, namely the oxygenated mycolic acids which are only found in pathogenic species, have been convincingly shown to be responsible for the LD accumulation in macrophages. On the other hand, non-virulent species only started to induce foamy macrophages after having been transformed by a gene that encodes an enzyme in charge of the oxygenation of mycolic acids.23 Apart from the infected macrophages, also non-infected neighboring macrophages accumulate LDs in infections by M. tuberculosis and M. leprae.4 This has been explained by the release of exosomes, consisting of microbial cell wall components, into the microenvironment. Subsequently, these exosomes stimulate the neighboring cells65 via certain toll-like receptors and induce LD formation. Given that co-stimulatory pro-inflammatory signals from tumor necrosis factor-alpha and/or monocyte chemo-tactic protein-1 are also present.4,90 it remains to be understood how this signaling results in increased gene expression of proteins such as acyl CoA synthase long chain family member 1and PLIN2.65
In the case of viral infections, alterations in lipid metabolism are notorious. Increased glucose uptake eventually leads to increased fatty acid biosynthesis as demonstrated by metabolic studies with [13C]glucose.74 The presence of fatty acid synthase is also essential for the production of infectious dengue viral particles; the NS3 protein localizes with this enzyme and redistributes it to viral replication sites.78 As mentioned before pharmacological inhibition of fatty acid synthesis or LD biogenesis diminishes virus replication. Furthermore, human CMv increases mRNA levels of phospholipase A2 and COX-2, and consistently arachidonic acid levels and PGE2 levels increase.79,91 The CMv immediate-early gene products, IE72 and IE84, can activate the COX-2 promoter via transactivation.92 The importance of COX-2 activity may be emphasized by the fact that the rhesus CMv genome encodes a COX-2 homologue.93 Furthermore, specific inhibitors of COX-2 not only block the accumulation of PGE2 after CMv infection but even reduce virus yields in cultured cells.92 Decreased viral replication upon treatment with the COX-2 inhibitor acetyl salicylic acid has also been reported for HCV in a subgenome in vitro model.94
Additionally, the NSAiD COX-2 inhibitors, tolfenamic acid and endomethacin, caused mislocalization of certain viral proteins, which remained inside the nucleus and did not reach the cytoplasm.95 The reduced production of infectious progeny could be restored when drug-blocked cultures were supplemented with PGE2, underscoring the essential function of PGE2 in efficient CMV replication.79,95
Conclusions
The field of lipidomics is rapidly growing and generating new data. LDs have received renewed interest in the area of infectious diseases. Certain intracellular pathogens induce LDs and take advantage of them by various means: a) some pathogens interfere in the regulation of lipid immunomodulators as a way of immunoevasion; b) others use the LD as a source of energy and essential nutrients, and even may establish themselves within the LD; c) some viruses use LD as assembly centers for infectious viral particles; and d) blood-feeding parasites use them in a detoxification process. Further investigation is required to better understand the induction and formation of LDs. Altered lipid metabolism and LD formation deserves attention in certain infectious diseases.
Acknowledgements
This work was supported by a grant to AGRT from CONACYT (CB-2008 Sol. 99149). IM received a scholarship from CONACYT No. 229067. The authors are grateful to Bonnie Young and Ossian Longoria-Lozano for reviewing the English.
Corresponding author: Dr. Mario César Salinas-Carmona.
Ave. Gonzalitos 235 Nte. Mitras Centro Monterrey, N. L. México. C.P. 64460.
Phone: +52 (81) 83294211, ext: 2794.
E-mail:msalinas@hu.uanl.mx
Recibido: Julio 2011.
Aceptado: Octubre 2011