


La enfermedad de Fabry es una enfermedad de depósito lisosomal causada por la deficiencia de la enzima alfa galactosidasa A, con patrón de herencia ligado al cromosoma X. Los avances en las últimas 2 décadas en cuanto a su diagnóstico, tratamiento y seguimiento han producido un mayor interés entre los profesionales de distintas especialidades, con el consiguiente aumento en el número de casos nuevos diagnosticados en la región. Este documento de revisión y consenso fue desarrollado por médicos de diversas especialidades de la región involucrados en la atención médica de pacientes con enfermedad de Fabry. La discusión y el análisis de la evidencia científica disponible a la fecha, sumados a la experiencia de cada uno de los participantes, fueron tomados en cuenta para unificar criterios y realizar recomendaciones con el propósito de brindar una herramienta útil para todos los profesionales que potencialmente se vean implicados en el diagnóstico, el tratamiento y el seguimiento de los pacientes en la región. La presente revisión abarca todos los aspectos discutidos en la última reunión de consenso del Grupo Centroamericano y del Caribe para el Estudio y Tratamiento de la Enfermedad de Fabry, incluyendo los objetivos del grupo, una introducción general a la enfermedad, las manifestaciones clínicas y el seguimiento recomendado por especialidad, cómo debe hacerse el diagnóstico y recomendaciones prácticas de manejo.
Fabry disease is a lysosomal storage disease caused by alpha galactosidase A enzyme deficiency. The pattern of inheritance is X-linked. The advances on its diagnosis, treatment and follow-up over the last two decades have led to a greater interest among professionals from different specialties, with the resulting increase in the number of newly diagnosed cases in the region. This review and consensus document was developed by physicians of many medical specialties and sub-specialties from the region directly responsible for the health care of patients with Fabry disease. Discussion and analysis of the available evidence, added to the experience of each of the participants, was considered in order to unify criteria and to make recommendations. The aim is to provide a valuable tool for every professional who may potentially become involved in the diagnosis, treatment, and follow-up of these patients in the region. This review covers all the aspects discussed during the last consensus meeting of the Central American and Caribbean Group for the Study and Treatment of Fabry disease, including its objectives, a general introduction to the disease, its clinical manifestations, and the recommended follow-up by the specialty, as well as how to make the diagnosis and clinical management recommendations.
Esta revisión es el producto de la reunión el Grupo Centroamericano y del Caribe para el Estudio y Tratamiento de la Enfermedad de Fabry, realizada en ciudad de Panamá, Panamá, del 14 al 16 de junio del 2016. Pretende difundir el conocimiento de los diferentes aspectos de la enfermedad de Fabry en la región centroamericana y del Caribe, desarrollando una acción integradora entre los diferentes componentes del equipo de salud involucrados que debe intervenir en el manejo de la enfermedad. Este grupo de referentes especialistas en pediatría, neurología, cardiología, nefrología, oftalmología, dermatología y otorrinolaringología, mediante la revisión de la evidencia disponible y la unificación de criterios para el diagnóstico, tratamiento y seguimiento en estos pacientes, considera este consenso como una herramienta para la educación médica continua, un documento de referencia y consulta para los colegas interesados en el manejo de esta enfermedad, así como también para las autoridades y agencias regulatorias encargadas de la cobertura de tratamientos de alto costo en los países de la región.
Objetivos- –
Realizar una revisión actualizada de la información científica disponible a la fecha y un intercambio de las experiencias acumuladas en nuestros países en el manejo de los pacientes con enfermedad de Fabry.
- –
Consensuar criterios de manejo clínico práctico, sobre la base de una evaluación de los recursos científicos, sanitarios, humanos y técnicos propios de la zona.
- –
Generar oportunidades de investigación y colaboración entre los colegas de nuestra región, para mejorar el manejo de nuestros pacientes.
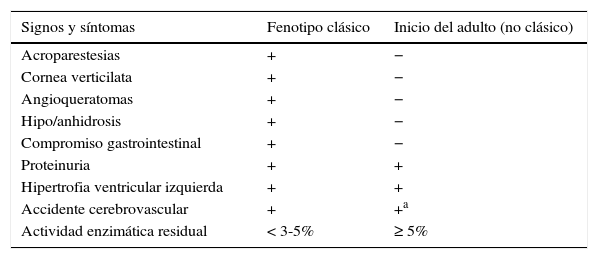
La enfermedad de Fabry es una enfermedad hereditaria de depósito lisosomal causada por mutaciones específicas en el gen GLA, localizado en el cromosoma X, que producen deficiencia o ausencia de la enzima alfa-galactosidasa A [1]. Se comporta como una enfermedad crónica, multisistémica y progresiva que deteriora la calidad de vida del paciente y reduce significativamente su sobrevida. Se caracteriza por el depósito de glicolípidos, principalmente globotriaosilceramida (Gb3 o GL-3) y globotriaosilesfingosina (liso-Gb3 o liso-GL-3), en diferentes tejidos y órganos, como endotelio vascular, nervios periféricos, piel, aparato digestivo, córnea y, con mayor severidad, en el sistema nervioso central, corazón y riñones [2]. Se han descrito 2 formas clínicas o fenotipos de la enfermedad: la forma clásica o severa y la forma de inicio en la edad adulta, también llamada variante no clásica o de inicio tardío, donde el daño se manifiesta principalmente en tejido renal o cardíaco, con complicaciones cerebrovasculares secundarias [1,2] (tabla 1).
Descripción de signos y síntomas con relación al fenotipo
| Signos y síntomas | Fenotipo clásico | Inicio del adulto (no clásico) |
|---|---|---|
| Acroparestesias | + | − |
| Cornea verticilata | + | − |
| Angioqueratomas | + | − |
| Hipo/anhidrosis | + | − |
| Compromiso gastrointestinal | + | − |
| Proteinuria | + | + |
| Hipertrofia ventricular izquierda | + | + |
| Accidente cerebrovascular | + | +a |
| Actividad enzimática residual | < 3-5% | ≥ 5% |
Es una de las enfermedades de depósito lisosomal más frecuentes. En su variante clásica, la incidencia es aproximadamente de 1 por cada 117.000 nacidos vivos y de 1 por cada 40.000 varones [1,3]. Durante la última década, los estudios de pesquisa neonatal y en poblaciones de riesgo, tales como en pacientes en tratamiento sustitutivo renal (terapia sustitutiva renal [TSR], diálisis, hemodiálisis o trasplante) o con miocardiopatía hipertrófica han mostrado que la variante del adulto puede ser mucho más frecuente, reportándose hasta en uno de cada 3.100 varones nacidos [4].
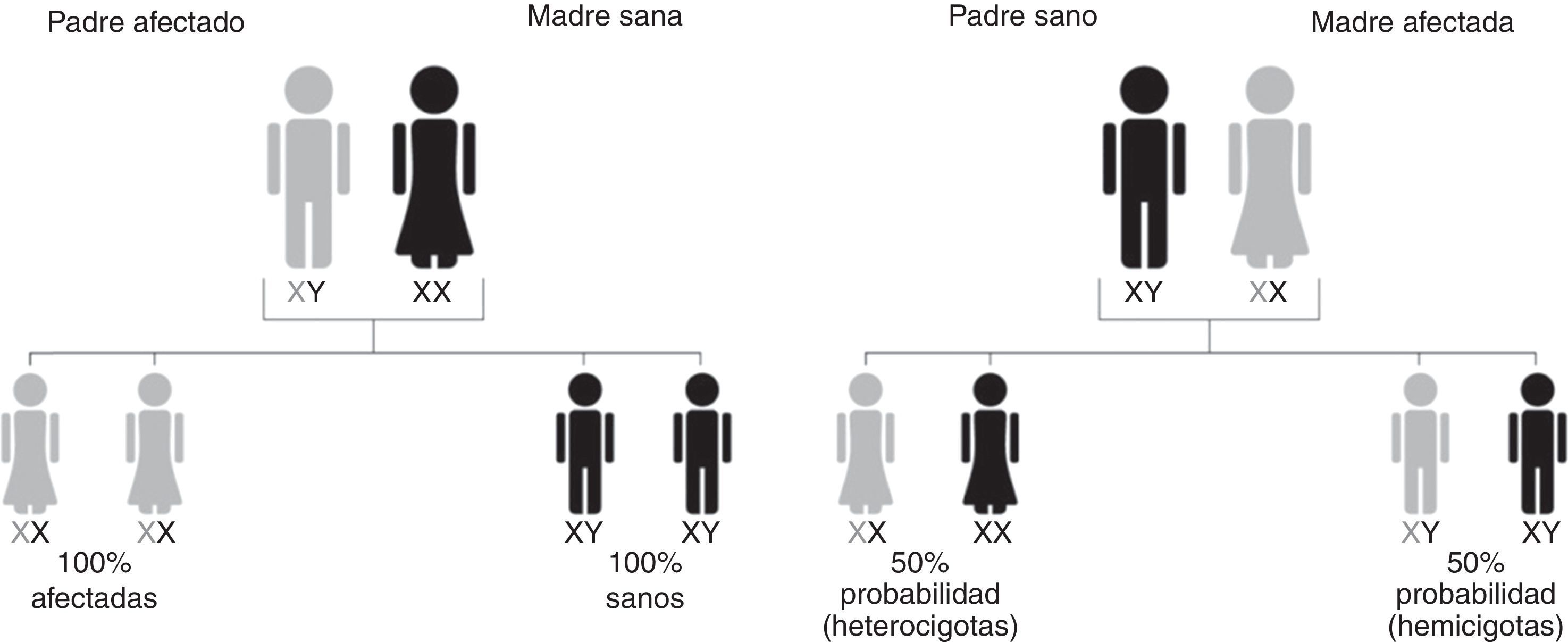
HerenciaSu transmisión está ligada al cromosoma X. El gen GLA que codifica para la enzima alfa galactosidasa se localiza en la banda Xq22.1 del brazo largo de dicho cromosoma [5]. En los varones (hemicigotos) se observa una alta penetrancia, aunque la expresión fenotípica del defecto enzimático presenta amplias variaciones intra e interfamiliares [6]. Las mujeres, consideradas antiguamente solo como portadoras, también pueden verse afectadas y en algunas ocasiones pueden presentar formas floridas de la enfermedad como consecuencia de la inactivación no aleatoria del cromosoma X sano, aunque los síntomas propios de la enfermedad pueden ser leves o estar ausentes [1,2] (fig. 1).

Patrón de herencia en la Enfermedad de Fabry. El gen responsable de la enfermedad de Fabry se localiza en el cromosoma X. Por la forma en que se hereda la enfermedad, los hombres, que tienen un único cromosoma X, no pueden transmitir la condición a sus hijos. Sin embargo, todas las hijas tendrán una copia del gen defectuoso. Las mujeres tienen 2 cromosoma X, uno de los cuales tendrá una copia del gen defectuoso. Las mujeres con enfermedad de Fabry tienen una probabilidad del 50% de transmitir el gen defectuoso a su descendencia, independientemente de si el hijo es niño o niña.
Antes del advenimiento del tratamiento farmacológico nefroprotector, del TSR y de la terapia de reemplazo enzimático (TRE), los pacientes con enfermedad de Fabry fallecían en promedio a los 41 años de edad, mayormente por enfermedad renal crónica (ERC) [7]. Actualmente, la edad promedio de muerte es a los 55 años en el 50% de los pacientes hemicigotos. Según un estudio reciente de bases de datos, actualmente la causa de muerte más frecuente es la cardiovascular, en el orden del 34% para varones y el 57% para mujeres [8]. Es importante notar que en dicho estudio la mayoría de los fallecidos habían sido diagnosticados de forma tardía y presentaban severo compromiso cardíaco y renal [8].
Diagnósticos diferencialesDado el amplio espectro de manifestaciones clínicas de la enfermedad de Fabry, es importante considerar algunos diagnósticos diferenciales como: esclerosis múltiple, artritis reumatoidea, fiebre reumática, enfermedad celíaca, síndrome de intestino irritable, intolerancia a la lactosa, porfiria, síndrome de Raynaud, fibromialgia, canalopatías dolorosas por mutaciones en genes que codifican proteínas para canales de sodio, nefroesclerosis hipertensiva, miocardiopatía hipertrófica familiar o idiopática y vasculitis cerebral [9].
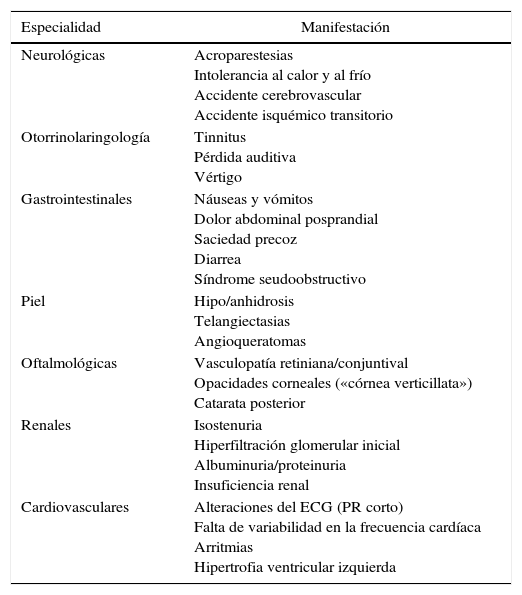
Manifestaciones clínicas y seguimiento por especialidadManifestaciones clínicasLa enfermedad de Fabry es un trastorno clínicamente heterogéneo que produce una amplia variedad de manifestaciones, por lo que el cuadro clínico puede ser muy diverso [1,2] (tabla 2). En la variante clásica es común encontrar desde la infancia dolores neuropáticos, angioqueratomas y córnea verticilada. Con el tiempo, los pacientes desarrollan ERC y lesiones cardíacas o de otros órganos, llegando habitualmente a necesitar TSR durante la cuarta década de la vida [7]. En la variante no clásica los pacientes pueden transcurrir completamente asintomáticos hasta la edad adulta, cuando se manifiestan las complicaciones graves, habitualmente renales y cardíacas [10]. Las manifestaciones clínicas requieren de un seguimiento periódico específico según la edad de los pacientes (tabla 3).
Manifestaciones de la Enfermedad de Fabry por especialidad
| Especialidad | Manifestación |
|---|---|
| Neurológicas | Acroparestesias Intolerancia al calor y al frío Accidente cerebrovascular Accidente isquémico transitorio |
| Otorrinolaringología | Tinnitus Pérdida auditiva Vértigo |
| Gastrointestinales | Náuseas y vómitos Dolor abdominal posprandial Saciedad precoz Diarrea Síndrome seudoobstructivo |
| Piel | Hipo/anhidrosis Telangiectasias Angioqueratomas |
| Oftalmológicas | Vasculopatía retiniana/conjuntival Opacidades corneales («córnea verticillata») Catarata posterior |
| Renales | Isostenuria Hiperfiltración glomerular inicial Albuminuria/proteinuria Insuficiencia renal |
| Cardiovasculares | Alteraciones del ECG (PR corto) Falta de variabilidad en la frecuencia cardíaca Arritmias Hipertrofia ventricular izquierda |
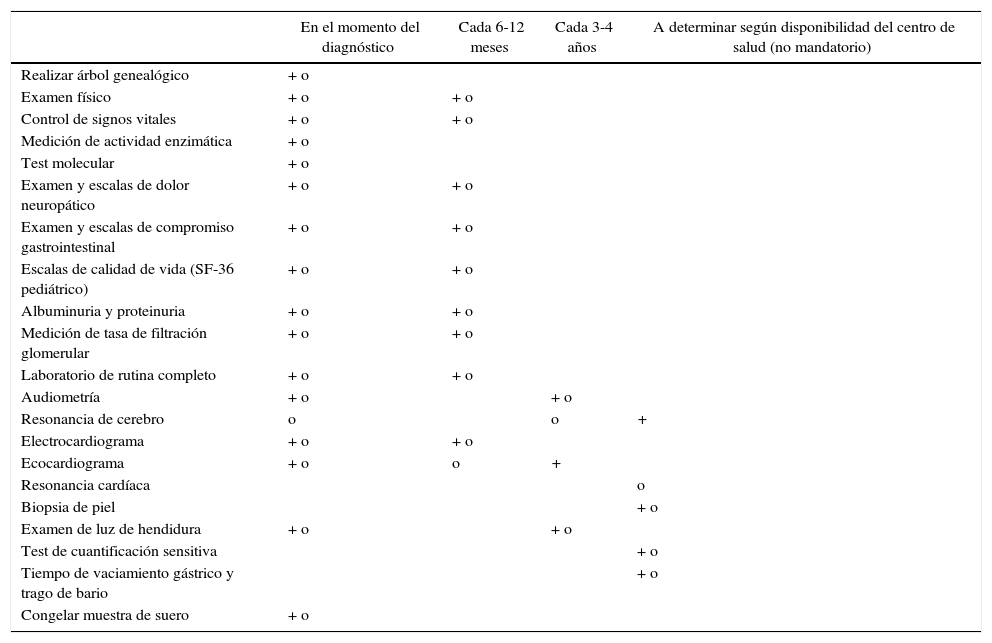
Estudios y evaluaciones recomendados para el seguimiento de los pacientes menores de 16 años (+) y adultos (o)
| En el momento del diagnóstico | Cada 6-12 meses | Cada 3-4 años | A determinar según disponibilidad del centro de salud (no mandatorio) | |
|---|---|---|---|---|
| Realizar árbol genealógico | + o | |||
| Examen físico | + o | + o | ||
| Control de signos vitales | + o | + o | ||
| Medición de actividad enzimática | + o | |||
| Test molecular | + o | |||
| Examen y escalas de dolor neuropático | + o | + o | ||
| Examen y escalas de compromiso gastrointestinal | + o | + o | ||
| Escalas de calidad de vida (SF-36 pediátrico) | + o | + o | ||
| Albuminuria y proteinuria | + o | + o | ||
| Medición de tasa de filtración glomerular | + o | + o | ||
| Laboratorio de rutina completo | + o | + o | ||
| Audiometría | + o | + o | ||
| Resonancia de cerebro | o | o | + | |
| Electrocardiograma | + o | + o | ||
| Ecocardiograma | + o | o | + | |
| Resonancia cardíaca | o | |||
| Biopsia de piel | + o | |||
| Examen de luz de hendidura | + o | + o | ||
| Test de cuantificación sensitiva | + o | |||
| Tiempo de vaciamiento gástrico y trago de bario | + o | |||
| Congelar muestra de suero | + o |
El dolor neuropático (acroparestesias) suele ser el síntoma inicial desde muy temprana edad y no responde a analgésicos comunes. El paciente puede describirlo como ardor, quemazón, electricidad u hormigueo bilateral en manos y/o pies, de intensidad variable desde moderado hasta crisis de dolor incapacitante [11]. Estas crisis se desencadenan típicamente durante el ejercicio, estados febriles o con los cambios en la temperatura ambiente. El examen neurológico puede ser normal al inicio. El electromiograma y las velocidades de conducción motoras y sensitivas son normales [12].
Debido al daño por acúmulo de Gb3 y liso-Gb3 en los ganglios submucosos y mientéricos, es muy frecuente que el paciente presente diarrea, dolor abdominal tipo cólico náuseas, saciedad temprana, vómitos y/o estreñimiento [13].
Manifestaciones tardíasLuego de la tercera década de vida pueden persistir el dolor neuropático y la disautonomía, manifestada como déficit en la vasorreactividad cerebral, síncopes y ortostatismo, además del compromiso del sistema nervioso central, manifestado en eventos cerebrovasculares isquémicos (transitorios o infartos) y/o hemorrágicos, habitualmente a partir de la cuarta década de la vida [14]. Este mecanismo de daño puede ser silente y diagnosticarse como hallazgo incidental en una resonancia magnética cerebral, con o sin dolicoectasia vertebro-basilar [14].
Tratamiento de soporte- –
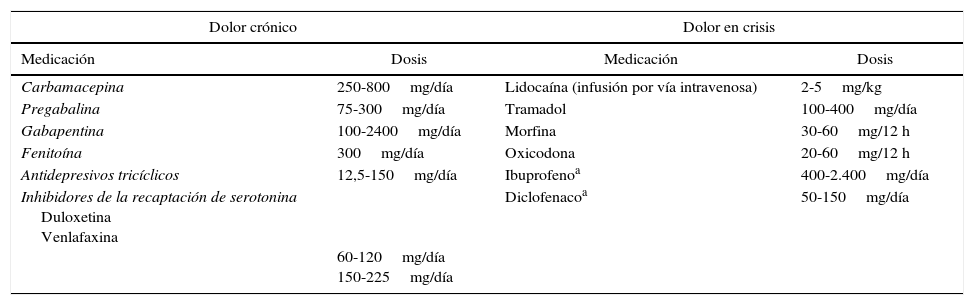
Medidas generales: tratamiento del dolor con analgésicos neuromoduladores (carbamazepina, gabapentina o pregabalina) (tabla 4).
Tabla 4.Tratamiento farmacológico recomendado para el tratamiento del dolor neuropático en la enfermedad de Fabry
Dolor crónico Dolor en crisis Medicación Dosis Medicación Dosis Carbamacepina 250-800mg/día Lidocaína (infusión por vía intravenosa) 2-5mg/kg Pregabalina 75-300mg/día Tramadol 100-400mg/día Gabapentina 100-2400mg/día Morfina 30-60mg/12 h Fenitoína 300mg/día Oxicodona 20-60mg/12 h Antidepresivos tricíclicos 12,5-150mg/día Ibuprofenoa 400-2.400mg/día Inhibidores de la recaptación de serotonina
Duloxetina
Venlafaxina
60-120mg/día
150-225mg/díaDiclofenacoa 50-150mg/día - –
En caso de hipoacusia súbita, pueden indicarse corticoides y vasodilatadores.
- –
De presentarse vértigo severo o incapacitante puede usarse betahistina.
- –
Para tratar los síntomas gastrointestinales se utilizan comúnmente antiespasmódicos, reguladores del tránsito y antidiarreicos.
- –
Se recomienda control y tratamiento de cualquier factor de riesgo cardiovascular concomitante: diabetes, obesidad, hipertensión arterial, dislipidemia y tabaquismo.
La cardiopatía se caracteriza por arritmias e hipertrofia ventricular izquierda (HVI), con posible progresión a insuficiencia cardíaca [15]. La HVI no es consecuencia del acúmulo de Gb3, sino del estímulo hipertrófico de este sustrato. De hecho, en los pacientes afectados el Gb3 representa solo el 1-2% de la masa cardíaca [16].
Manifestaciones tempranasSuelen aparecer antes de los 35 años en forma de trastornos de la conducción como bradicardia, bloqueos de rama o acortamiento del intervalo PR. También puede observarse HVI, generalmente asintomática [17].
Manifestaciones tardíasSíntomasLuego de la tercera década de vida puede haber cuadros anginosos asociados a disnea, palpitaciones y mareos. La disautonomía cardíaca es común en esta etapa.
SignosLa HVI condiciona disfunción diastólica, progresando luego a fallo sistólico y fibrosis endomiocárdica [16]. Los trastornos de conducción y alteraciones de la repolarización ventricular izquierda se vuelven más evidentes y es posible la aparición de arritmias malignas que requieren marcapasos o cardiodesfibrilador implantable, según indicación. Puede haber edemas en los miembros inferiores como consecuencia de insuficiencia cardíaca [17].
Tratamiento de soporteEvaluar la necesidad de iniciar el TRE de forma temprana antes del desarrollo de fibrosis miocárdica, pues una vez establecida la fibrosis el compromiso cardíaco es irreversible y no responderá aún con TRE [18].
Si hay hipertensión arterial, debe ser tratada, idealmente con un inhibidor de la enzima conversora de angiotensina o con un análogo de los receptores de angiotensina II, si hay albuminuria. Las arritmias deben tratarse con fármacos específicos y la fibrilación auricular puede requerir de cardioversión eléctrica, según indicación [17].
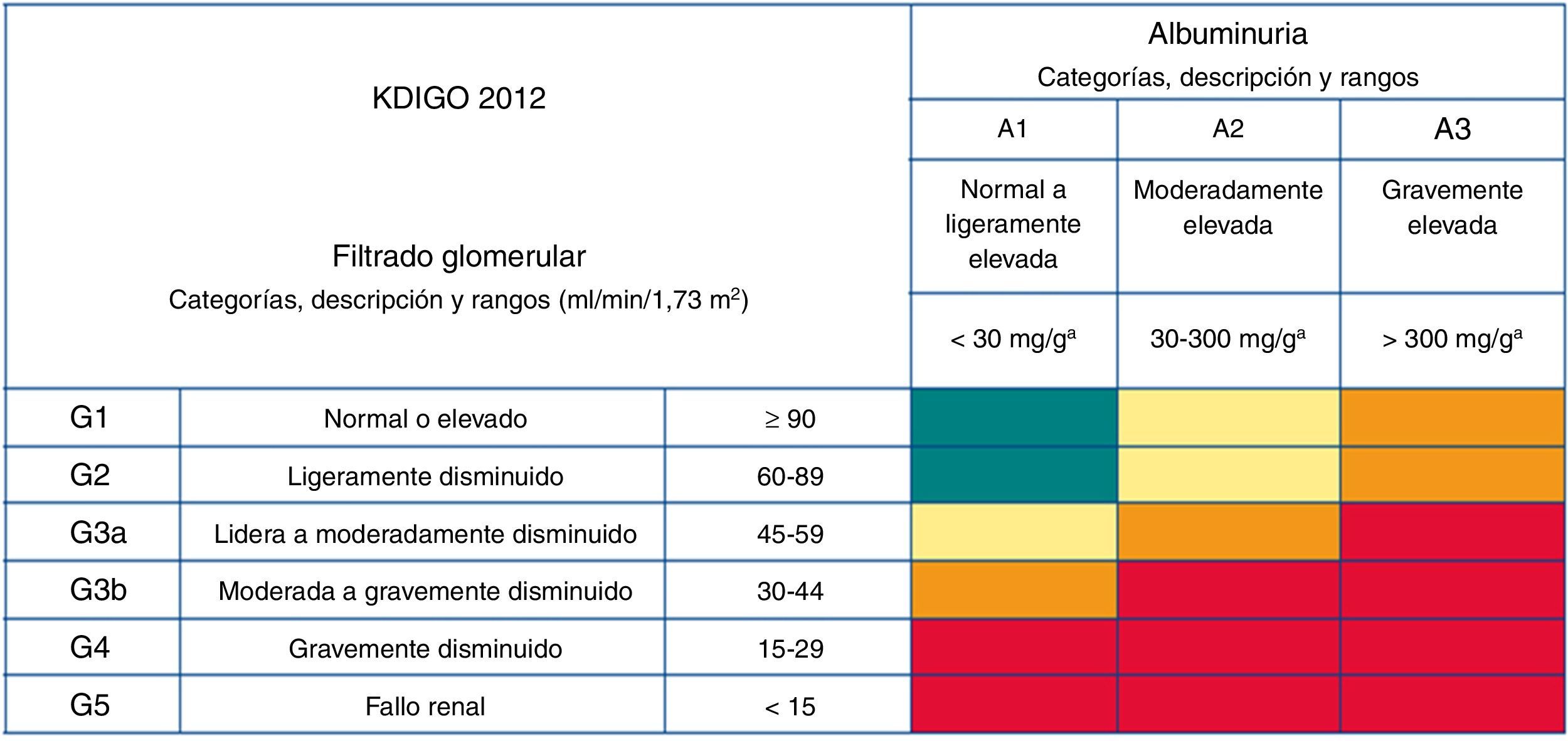
NefrologíaLa nefropatía en la enfermedad de Fabry se caracteriza por la presencia de albuminuria/proteinuria y progresa lenta y paulatinamente a ERC estadio 5 terminal con necesidad de TSR crónica en muchos pacientes, especialmente en quienes presentan de mutaciones no clásicas de manifestación tardía [19]. La progresión es más frecuente en varones, aunque algunas mujeres también llegan a requerir TSR [20]. La mitad de los pacientes varones con fenotipo clásico presentarán compromiso renal antes de los 35 años y todos después de los 50 [21]. La función renal debe clasificarse por estadios de la tasa de filtración glomerular (TFG) y por categorías de albuminuria según las guías Kidney Disease: Improving Global Outcomes (KDIGO) [22] (fig. 2). Para estimar la TFG, se recomienda utilizar la fórmula CKD-EPI [23] en adultos y la fórmula modificada de Schwartz [24] en niños.
 de las guías Kidney Disease: Improving Global Outcomes (KDIGO), definidas mediante categorías de tasa de filtración glomerular estimada (TFGe) y de albuminuria, para establecer niveles de riesgo. Verde: Bajo riesgo (no es ERC si no hay otros marcadores de ERC); amarillo: riesgo moderadamente incrementado; naranja: alto riesgo; rojo: muy alto riesgo. Modificado y traducido al español de Kidney Disease: Improving Global Outcomes (KDIGO) CKD working group. KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Int Suppl. 2013;3(1):1-150.")
Estadios de la Enfermedad renal crónica (ERC) de las guías Kidney Disease: Improving Global Outcomes (KDIGO), definidas mediante categorías de tasa de filtración glomerular estimada (TFGe) y de albuminuria, para establecer niveles de riesgo.
Verde: Bajo riesgo (no es ERC si no hay otros marcadores de ERC); amarillo: riesgo moderadamente incrementado; naranja: alto riesgo; rojo: muy alto riesgo.
Modificado y traducido al español de Kidney Disease: Improving Global Outcomes (KDIGO) CKD working group. KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Int Suppl. 2013;3(1):1-150.
El comportamiento clínico es el de una glomerulopatía.
- –
En estadios de ERC tempranos la lesión puede manifestarse inicialmente como hiperfiltración con o sin albuminuria. En estadios posteriores, la albuminuria se vuelve más franca y la TFG declina paulatinamente hasta llegar a ERC estadio 5 [19].
- –
Se ha descrito la presencia de albuminuria desde muy temprana edad, incluso desde los 6 años, en algunos hemicigotos con fenotipo clásico [25].
- –
La albuminuria es de rango variable y es la expresión más frecuente de lesión renal. Sin embargo, puede haber lesión renal sin proteinuria en el 10% de los varones y el 30% de las mujeres con ERC estadio 3 [26].
- –
Si bien la albuminuria puede ser categoría A3 [22] y llegar a rango nefrótico en el 20% de los pacientes, rara vez se observa síndrome nefrótico [20].
- –
La podocituria, detectada mediante inmunofluorescencia, es un biomarcador aún más precoz de lesión renal [27], pero esta técnica no se encuentra todavía disponible en muchos centros de la región.
- –
Menos del 20% de los pacientes presentan hematuria microscópica [20].
- –
Como hallazgo semiológico, algunos pacientes pueden presentar edema de grado variable y orina turbia y espumosa.
Es muy importante mencionar que la biopsia renal no es imprescindible para el diagnóstico de la enfermedad de Fabry, ya que en la mayoría de los pacientes otros signos y síntomas suelen presentarse de forma más precoz que la lesión renal [28]. Sin embargo, la biopsia puede ser de utilidad y debe considerarse en situaciones particulares. De realizarse, es necesario utilizar tinción de hematoxilina-eosina y azul de toluidina en el estudio de microscopia de luz y se recomienda guardar una muestra de tejido en glutaraldehído para practicar microscopia electrónica, si está disponible [28]. Se recomienda realizar biopsia renal para descartar patología concomitante en:
- –
Pacientes previamente estables que presenten un deterioro rápido de la función renal o una caída súbita de la TFG.
- –
Pacientes previamente estables que presenten un incremento súbito de la albuminuria, o que presenten albuminuria categoría A3 o proteinuria en rango nefrótico.
- –
Pacientes con síndrome nefrítico o nefrótico.
- –
Cuando se presente una situación de duda diagnóstica en la cual se requiera valorar la presencia y el grado de lesión renal, como, por ejemplo, en pacientes mujeres en quienes el hallazgo de compromiso en los podocitos puede tener impacto en el pronóstico y en la decisión terapéutica [29].
Dependiendo de la función renal, determinada por la TFG y la categoría de albuminuria, deben seguirse las medidas generales de nefroprotección establecidas en las guías KDIGO que incluyen, pero no se limitan a: una adecuada hidratación, el uso de medicamentos nefroprotectores y el control de los factores de riesgo de progresión como la hipertensión arterial y anemia [22].
En pacientes con ERC estadio 5 el TSR está indicado en cualquiera de sus modalidades (diálisis peritoneal, hemodiálisis o trasplante). La enfermedad de Fabry no contraindica el trasplante, aunque deben evaluarse adecuadamente los otros órganos posiblemente afectados, particularmente el corazón.
En dosis adecuadas, la TRE ha demostrado disminuir la velocidad de progresión de la ERC [30]. La TRE debe continuarse —o puede iniciarse— en pacientes que ya se encuentran en TSR con el fin de proteger otros órganos, principalmente corazón y el cerebro, para evitar mayores complicaciones o eventos fatales.
DermatologíaManifestaciones clínicas- 1.
Angioqueratomas
- –
Aparecen desde la infancia o adolescencia. Por lo general se encuentran agrupados en la región glútea, la región umbilical, muslos y genitales [1].
- –
Los angioqueratomas diseminados son altamente indicativos de enfermedad de Fabry pero no patognomónicos. Puede realizarse biopsia de piel para descartar diagnósticos diferenciales de las lesiones [1,2].
- –
El estudio anatomopatológico de la biopsia de piel muestra dolicoectasia de vasos de la dermis con microtrombosis en su interior. La ausencia de queratosis suele describirse en algunas lesiones, que continúan siendo típicas de la enfermedad [31].
- –
- 2.
Telangiectasias en piel y mucosas
- –
Se observan predominantemente en rostro, labios y mucosa oral.
- –
- 3.
Alteraciones en la sudoración
- –
La hipohidrosis o anhidrosis es frecuente y suele ser un signo precoz en la infancia, provocando piel seca, intolerancia al calor o al ejercicio y fiebre de origen desconocido. También puede disminuirse la producción de lágrimas y saliva [1].
- –
- 4.
Otras manifestaciones menos frecuentes son: linfedema, disminución del vello corporal y alopecia difusa [32].
- 1.
Las lesiones dermatológicas deben biopsiarse siempre pues no toda neoformación roja es angioqueratoma y el diagnóstico diferencial puede ir desde un nevo rubí hasta melanomas.
- 2.
No es frecuente que las lesiones dermatológicas involucionen con el TRE.
- 3.
Los angioqueratomas pueden tratarse con terapia de luz pulsada intensa. No siempre hay involución completa, pero sí notable mejoría [33].
- 4.
La electrocirugía es la mejor opción para las lesiones sangrantes. El uso de sedoanalgesia no está contraindicado.
- 1.
Diagnósticas
- –
Córnea verticilada (útil para el diagnóstico, aunque no compromete la visión).
- –
Tortuosidad vascular conjuntival y retinal (vasos en omega).
- –
Catarata subcapsular posterior.
- –
- 2.
Morbilidad
- –
Los fenómenos oclusivos retinianos y la ceguera no son frecuentes.
- –
- 1.
Ojo externo
- –
Búsqueda de alteraciones vasculares conjuntivales: presencia/ausencia de tortuosidad vascular, presencia/ausencia de telangiectasias [1].
- –
- 2.
Examen con lámpara de hendidura
- –
Oresencia/ausencia de córnea verticilada: presencia/ausencia de cataratas [2].
- –
- 3.
Examen de fondo de ojos
- –
Presencia/ausencia de tortuosidad vascular (arteria/venosa).
- –
Son el resultado del compromiso del oído interno, tanto del laberinto anterior (audición) como del posterior (equilibrio), y de la vía neurológica auditiva (octavo par craneal, tronco cerebral y corteza auditiva).
- 1.
Hipoacusia
- –
Tipo perceptiva o neurosensorial, unilateral o bilateral, progresiva o de instalación súbita (en el lapso de 72 h), de grado leve a severa [34]. No presenta un perfil audiométrico patognomónico y puede comprometer las frecuencias agudas o ser pantonal (todas las frecuencias).
- –
- 2.
Acúfenos
- –
Unilaterales o bilaterales, esporádicos o permanentes, de tonalidad aguda [1].
- –
- 3.
Vértigo
- –
Se observa en crisis espontáneas de breve duración.
- –
Está asociado a la pérdida auditiva.
- –
Se presenta con sordera súbita en frecuencias agudas.
- –
- 4.
Ataxia y oscilopsia por compromiso vestibular bilateral, con o sin compromiso auditivo [34].
Audiometría.
Potenciales evocados auditivos.
Potenciales miogénicos vestibulares.
Tratamiento de soporteSi aparece hipoacusia o vértigo súbito, se recomienda el tratamiento habitual de urgencia inespecífico: corticoterapia, vasodilatadores y antiagregantes plaquetarios.
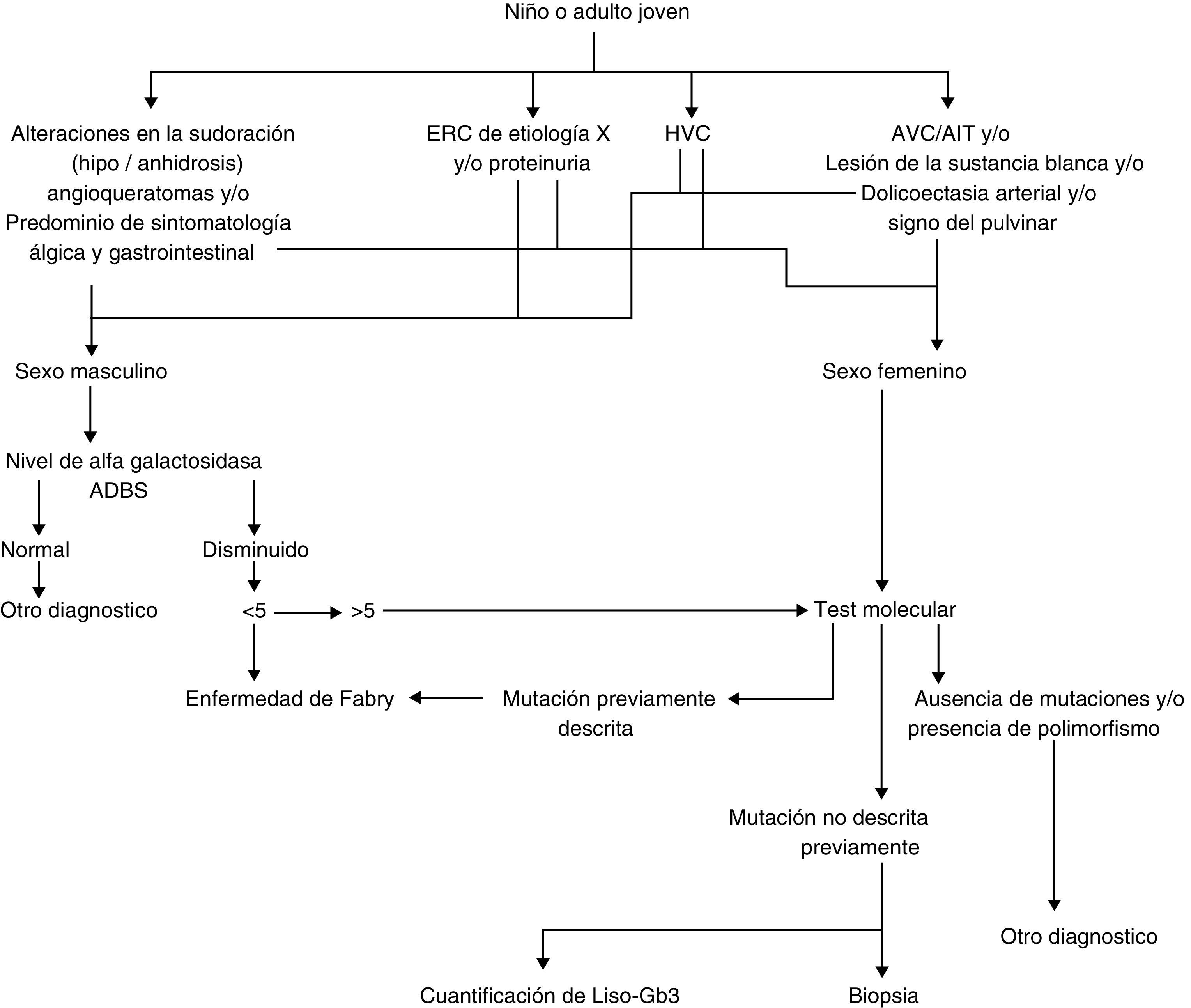
¿Cómo debe hacerse el diagnóstico? (fig. 3)Actividad enzimática de alfa-galactosidasa AEl diagnóstico definitivo de la enfermedad de Fabry en varones con fenotipo clásico se establece al determinar deficiencia o ausencia de la actividad de la alfa-galactosidasa A en leucocitos aislados de sangre periférica [35]. La determinación enzimática puede también realizarse en gotas de sangre seca en papel filtro. Esto permite el envío de muestras a distancia, el diagnóstico retrospectivo y el tamizaje poblacional [36]. No obstante, un resultado anormal en la prueba del papel filtro debe confirmarse con la prueba en leucocitos o mediante estudio molecular (test genético) [35]. En las mujeres, debido a la inactivación aleatoria de uno de los cromosomas X, la actividad enzimática de la alfa-galactosidasa A puede ser normal hasta en el 40% de los casos (falso negativo). Por tanto, un valor normal de actividad enzimática no descarta la enfermedad en mujeres y para el diagnóstico debe hacerse estudio genético [37]. Es importante considerar que la medición de la actividad enzimática también puede producir falsos positivos, es decir, puede haber actividad enzimática disminuida en personas sanas portadoras de variantes genéticas de significado incierto o de polimorfismos no patogénicos [38]. En estos pacientes la sola demostración de valores de actividad enzimática disminuida no es indicación de inicio de TRE sin haber realizado antes una confirmación molecular o, en casos excepcionales, una determinación de biomarcadores en plasma o tejidos mediante biopsia. El estudio de la actividad enzimática es útil solamente como apoyo diagnóstico y no debe utilizarse para el seguimiento del paciente o para medir la eficacia de la TRE.
Diagnóstico prenatal
El diagnóstico prenatal puede hacerse en muestras de vellosidades coriónicas obtenidas a partir de la semana 11 de gestación o en cultivo de células del líquido amniótico a partir de la semana 16 [2]. Todo estudio prenatal debe ser confirmado luego del parto en el recién nacido.
Diagnóstico molecularDebe identificarse cualquiera de las múltiples mutaciones responsables de la enfermedad. En varones con fenotipo clásico, el estudio de la secuencia codificante del gen GLA para identificar la mutación patogénica es una prueba diagnóstica complementaria. La identificación de la mutación es particularmente útil para el estudio familiar y constituye la prueba confirmatoria definitiva en mujeres sintomáticas con actividad enzimática normal [35]. Algunas técnicas utilizadas en la práctica diaria para detectar cambios en el gen GLA pueden ser no concluyentes en algunos casos, ya que en un pequeño porcentaje algunas mutaciones (inversiones, duplicaciones, o mutaciones intrónicas o en el promotor) requieren de técnicas más complejas para su detección, evitando así falsos negativos. El estudio molecular o genético no debe utilizarse para seguimiento de la efectividad del TRE, su utilidad se limita al diagnóstico.
Niveles de biomarcadores: Gb3 y liso-Gb3Tanto el Gb3 como el liso-Gb3 se encuentran elevados en plasma y en orina en pacientes varones con enfermedad de Fabry clásica [39,40], pero pueden estar normales o solo ligeramente aumentados en pacientes con variantes de inicio del adulto o en mujeres (heterocigotas). La determinación de estos metabolitos no debe realizarse de forma rutinaria, sino excepcionalmente cuando sea necesario. Otra forma de confirmar la presencia por acúmulo patológico de estos biomarcadores es la demostración de inclusiones de sustrato en tejidos por medio de biopsia de conjuntiva, piel, riñón o corazón [41].
Tratamiento específicoDesde el año 2001 se dispone de TRE con alfa-galactosidasa A como tratamiento específico para la enfermedad de Fabry. Otros tratamientos están siendo evaluados con resultados preliminares prometedores (terapia génica, inhibición del sustrato, etc.) y el uso de chaperonas químicas, una nueva clase de pequeñas moléculas que actúan mediante la estabilización de proteínas inestables, ya está aprobado en Europa (clorhidrato de migalastat) para el uso en Fabry, pero de momento la TRE es el único tratamiento específico disponible en la región.
Terapia de reemplazo enzimáticoSu principal mecanismo de acción es la sustitución de la enzima alfa-galactosidasa A, posibilitando la hidrólisis del Gb3 y liso-Gb3 al separar un residuo de galactosa terminal de la molécula, disminuyendo así su acumulación en los tejidos [1].
Existen 2 formulaciones de TRE: la agalsidasa alfa y la agalsidasa beta. Ambas han sido aprobadas en Europa y Latinoamérica, aunque en Estados Unidos de América la Food and Drug Administration solamente ha aprobado la agalsidasa beta desde 2003 hasta la fecha [42,43]. Ambas TRE están compuestas por la misma cadena de aminoácidos, es decir, son la misma proteína humana. La diferencia reside en la dosis aprobada (agalsidasa beta 1mg/kg de peso y agalsidasa alfa 0,2mg/kg) y en la cantidad de residuos de manosa 6-fosfato (agalsidasa beta 3,6mol/mol de proteína y agalsidasa alfa 1,3mol/mol de proteína) [44]. La presencia de mayor cantidad de residuos de manosa 6-fosfato produce un mejor reconocimiento por los receptores de manosa 6-fosfato de la membrana celular y consecuentemente mayor internalización celular de la enzima [44]. De acuerdo con los estudios publicados, pareciera que el beneficio tisular y clínico tiene relación directa con la dosis de TRE utilizada [45–48]. Distintos protocolos han demostrado que el uso de agalsidasa beta a dosis de 1mg/kg cada 14 días produce remoción total de Gb3 de los podocitos renales en comparación con la agalsidasa alfa a dosis de 0,2mg/kg. Más aún, la reducción de la dosis de 1mg/kg a 0,2mg/kg cada 14 días conduce a reacúmulo del sustrato en los vasos y podocitos luego de varios años, reapareciendo también los síntomas de la enfermedad que habían sido resueltos con el uso de dosis mayores [49]. Por tanto, es recomendable utilizar la agalsidasa beta en la dosis aprobada de 1mg/kg cada 14 días. Durante la infusión de TRE pueden presentarse reacciones adversas que deben manejarse oportunamente (ver Anexo).
¿Cuándo debe iniciarse la terapia de reemplazo enzimático?Siguiendo las guías internacionales [50,51], está indicado iniciar TRE en todo paciente con enfermedad de Fabry confirmada (fenotipo clásico) que presente signos y síntomas característicos, sin distinción de edad ni sexo. Esta recomendación incluye las manifestaciones leves, que de igual forma son indicadores de daño tisular activo [52]. No es recomendable sustituir la TRE por tratamientos de soporte sintomáticos debido a que la resolución temporal de un síntoma, sin tratar el mecanismo fisiopatológico subyacente, resultará a largo plazo en mayor daño con escasa posibilidad de reversión o estabilización [50].
Los signos, los síntomas y las condiciones que en la práctica diaria llevan a iniciar la TRE son:
- –
Dolor neuropático distal en los 4 miembros.
- –
Intolerancia al calor/ejercicio y anhidrosis/hipohidrosis.
- –
Compromiso gastrointestinal (dolor abdominal recurrente postprandial, náuseas, vómitos y diarrea).
- –
Presencia de albuminuria categoría A1 y/o proteinuria.
- –
Depósitos de Gb3 con borramiento de los procesos podocitarios en la biopsia renal, aún previo al desarrollo de albuminuria/proteinuria.
- –
Arritmias y/o HVI.
- –
Evento cerebrovascular o evento isquémico transitorio.
Los pacientes varones asintomáticos que surgen de la pesquisa familiar luego del hallazgo de un paciente (caso índice) que presentan una mutación ligada al fenotipo clásico pueden recibir TRE precoz y no deberían esperar superar los 8 o 10 años de edad, aunque esta indicación está sujeta al criterio del especialista [50,51]. En el ámbito nefrológico el tratamiento es más efectivo si se inicia antes de presentarse deterioro de la función renal. La infusión de TRE puede presentar reacciones adversas que deben manejarse oportunamente (ver Anexo).
No inicio o interrupción del tratamientoSe sugiere valorar no iniciar o descontinuar el TRE en las siguientes situaciones:
- –
Pacientes severamente comprometidos y de pobre pronóstico, por ejemplo, pacientes con ERC estadio 5 sin posibilidad de trasplante, con insuficiencia cardíaca severa (NYHA clase IV) y deterioro cognitivo vascular isquémico.
- –
Falta de adherencia al TRE.
- –
Solicitud del paciente de descontinuar el TRE.
- –
Reacciones alérgicas severas a repetición al TRE que no responden al tratamiento con premedicación.
La enfermedad de Fabry es poco frecuente y heterogénea. Los avances en las técnicas de diagnóstico y la posibilidad de un tratamiento específico deben incrementar el índice de sospecha en la comunidad médica. En su diagnóstico y tratamiento intervienen múltiples especialistas, por lo que se recomienda un enfoque multidisciplinario.
Dado que la TRE ha demostrado cambiar la historia natural de la enfermedad, este grupo de especialistas intenta mejorar las condiciones de diagnóstico, tratamiento y monitorización de estos pacientes mediante la redacción de este documento.
Esta revisión fue realizada sobre la base de la literatura existente a la fecha y está sujeta a modificación, si se presenta nueva evidencia.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
FinanciaciónNo se recibió financiamiento para la redacción de esta revisión.
Conflicto de interesesTodos los autores son miembros del Grupo Centroamericano y del Caribe para el Estudio y Tratamiento de la Enfermedad de Fabry. La reunión del Grupo en junio de 2016 en Ciudad de Panamá, República de Panamá, fue patrocinada por Sanofi Genzyme. El patrocinador de la reunión del Grupo no ha participado en la redacción de este artículo ni ha tenido acceso a su contenido antes de su publicación.
AgradecimientosLos autores agradecen a Juan Manuel Politei, por su valioso aporte y revisiones durante la elaboración del manuscrito.
Medicación antes del TRE (premedicación), 1 h preinfusión:
- –
Paracetamol, 2 tabletas de 500mg por vía oral.
- –
Hidroxicina 1 tableta de 25mg por vía oral u otro antihistamínico similar, según disponibilidad local.
- –
Control de signos vitales (presión arterial, frecuencia cardíaca y respiratoria, temperatura).
- –
En caso de que el paciente presente fiebre o escalofríos, en las siguientes infusiones reemplazar el paracetamol por ibuprofeno 1 tableta de 400mg por vía oral.
Manejo de reacciones de hipersensibilidad
A. Reacciones leves, definidas como la presencia de:
- –
Sensación de calor.
- –
Urticaria menor o localizada (< 5% de superficie corporal).
- –
Hormigueos periféricos.
- –
Prurito cutáneo.
Conducta recomendada:
- 1.
Disminuir la velocidad de infusión a la mitad (registrar hora y velocidad de goteo).
- 2.
Administrar difenhidramina 50mg, por vía oral o intravenosa, en 5-10 min.
- 3.
Si los síntomas desaparecen, volver a la velocidad de infusión previa en forma escalonada (en 15-30 min).
- 4.
Si la severidad de los síntomas aumenta, detener infusión.
B. Reacciones moderadas, definidas como la presencia de:
- –
Disnea leve.
- –
Urticaria generalizada.
- –
Náuseas y vómitos.
- –
Taquicardia.
- –
Prurito generalizado, calor y ansiedad.
- –
Enrojecimiento (flushing).
- –
Angioedema.
Conducta recomendada:
- 1.
Detener la infusión para evaluar la severidad o alternativamente disminuir la velocidad de infusión a la mitad.
- 2.
Difenhidramina 50mg, por vía oral o intravenosa.
- 3.
Si hay síntomas respiratorios, utilizar beta-2 agonistas inhalados (salbutamol).
- 4.
Si persisten síntomas respiratorios, administrar adrenalina 1:1.000, 0,30-0,50ml por vía subcutánea en la extremidad superior (contraindicado en cardiópatas).
- 5.
Hidrocortisona 100mg por vía intravenosa y/o antipiréticos.
- 6.
Detener la infusión si los síntomas aumentan en severidad o persisten.
C. Reacciones severas, definidas como la presencia de:
- –
Disnea severa.
- –
Obstrucción de vía aérea.
- –
Arritmias.
- –
Hipotensión.
- –
Colapso cardiovascular.
Conducta recomendada:
- 1.
Detener inmediatamente la infusión.
- 2.
Administrar adrenalina 1:1.000, 0,30-0,50ml por vía subcutánea en la extremidad superior.
- 3.
Difenhidramina 50mg por vía intravenosa.
- 4.
Hidrocortisona 100mg por vía intravenosa.
- 5.
Para síntomas respiratorios utilizar beta-B2 agonistas inhalados o en nebulización.
- 6.
Si hay disnea severa con cianosis o sibilancias, colocar oxígeno por máscara o bigotera.
- 7.
Manejar volumen de fluidos.
- 8.
En caso de paro cardiorrespiratorio, iniciar maniobras de reanimación cardiopulmonar.