



Autoimmune autonomic ganglionopathy (AAG) is an acquired immune-mediated disorder.1 It was initially described in 1974 as pure pandysautonomia, and considered a variant of Guillain-Barré syndrome.2 In 1994, patients with the condition were found to present antibodies specific for the alpha-3 subunit of the ganglionic acetylcholine (ACh) receptor.3 Most cases are acute or subacute and occur in the context of recent history of respiratory tract infection.2 We present the case of a patient with slowly progressive dysautonomia and provide data from a small fibre study.
Our patient was a 43-year-old man with no relevant medical history. Approximately a year before the initial assessment, he began to present slowly progressive symptoms of orthostatic intolerance, with dizziness, blurred vision upon standing, and leg weakness when walking. Symptoms improved in the decubitus position. He subsequently began to present frequent syncopes. A cardiological evaluation ruled out heart disease, and the tilt test revealed orthostatic hypotension. The patient underwent an autonomic function study. He reported nocturia, erectile dysfunction, eye and mouth dryness, lack of sweating during exercise, and photophobia. A neurological examination revealed normal cognitive function. The cranial nerve examination showed bilateral fixed mydriasis, normal ocular motility, normal fifth and twelfth cranial nerve function, and normal motor and sensory function. The autonomic function study revealed alterations in cardiac sympathetic, cardiovagal, sudomotor, and pupillary function (Table 1). A blood test did not detect anaemia or leukocytosis, and showed normal erythrocyte sedimentation rate and normal levels of glucose, creatinine, vitamin B12, and folic acid. The patient tested negative for HIV infection and presented normal levels of serum gamma globulins. An immunity study revealed no anti-Ro, anti-La, or antinuclear antibodies. Imaging studies revealed no signs of tumour. Motor and sensory nerve conduction studies yielded normal results. A skin biopsy revealed normal density of unmyelinated fibres in the epidermis and dermis (sweat glands).
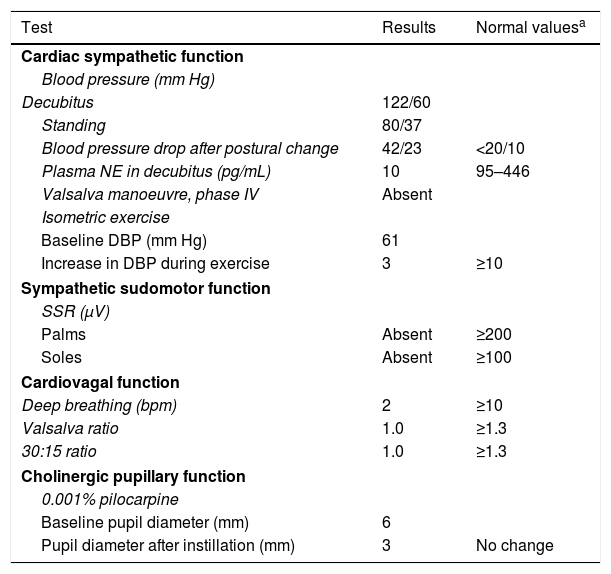
Autonomic function study results.
| Test | Results | Normal valuesa |
|---|---|---|
| Cardiac sympathetic function | ||
| Blood pressure (mm Hg) | ||
| Decubitus | 122/60 | |
| Standing | 80/37 | |
| Blood pressure drop after postural change | 42/23 | <20/10 |
| Plasma NE in decubitus (pg/mL) | 10 | 95–446 |
| Valsalva manoeuvre, phase IV | Absent | |
| Isometric exercise | ||
| Baseline DBP (mm Hg) | 61 | |
| Increase in DBP during exercise | 3 | ≥10 |
| Sympathetic sudomotor function | ||
| SSR (μV) | ||
| Palms | Absent | ≥200 |
| Soles | Absent | ≥100 |
| Cardiovagal function | ||
| Deep breathing (bpm) | 2 | ≥10 |
| Valsalva ratio | 1.0 | ≥1.3 |
| 30:15 ratio | 1.0 | ≥1.3 |
| Cholinergic pupillary function | ||
| 0.001% pilocarpine | ||
| Baseline pupil diameter (mm) | 6 | |
| Pupil diameter after instillation (mm) | 3 | No change |
DBP: diastolic blood pressure; NE: norepinephrine; SSR: sympathetic skin response.
Differential diagnosis of this progressive sympathetic and parasympathetic dysautonomia without sensory or motor involvement included Sjögren syndrome, which may present as dysautonomia associated with small or large sensory fibre involvement4; our patient did not present antibodies specific for this condition or sensory neuropathy. Amyloidosis may also present in the form of dysautonomia associated with peripheral axonal polyneuropathy.5 However, our patient did not present heart, kidney, or peripheral nerve involvement. We also considered paraneoplastic dysautonomia, which presents as acute or subacute symptoms of gastrointestinal pseudo-obstruction either in isolation or in association with encephalitis or sensory ganglionopathy.6 However, our patient did not present brain or peripheral nerve involvement, and no alterations were found in a chest, abdomen, and prostate examination. Another possible diagnosis was pure autonomic failure (PAF), which manifests as slowly progressive sympathetic and parasympathetic dysfunction without motor or sensory involvement, secondary to a chronic neurodegenerative process (synucleinopathy).7
The patient presented bilateral fixed mydriasis secondary to cholinergic denervation of the pupils, a frequent finding in cases of AAG with high antibody titres but rare in patients with PAF,8 which led us to request a study of ganglionic anti-ACh receptor antibodies. The patient tested positive for antibodies against the alpha-3 subunit of the ganglionic ACh receptor, at a concentration above 3.6 nmol/L (normal level, <0.05 nmol/L; Southwestern University, TX, USA). Orthostatic hypotension was treated with midodrine and fludrocortisone. Our patient received immunosuppressive therapy with prednisone and azathioprine; in subsequent months, orthostatic intolerance improved moderately, with occasional episodes of syncope. At a 4-year follow-up consultation, the patient reported no further episodes of syncope and no additional symptoms.
The main manifestations of AAG include orthostatic hypotension, constipation, severe pupillary cholinergic symptoms, urinary symptoms, dry eyes and mouth, and intolerance to heat due to anhidrosis.1,2 Antibodies block ganglionic transmission, causing dysautonomia.1 Treatment of acute AAG is based on intravenous immunoglobulins or plasmapheresis; long-term treatment includes prednisone, mycophenolate, azathioprine, and rituximab, in monotherapy or in combination.1,2 As our patient showed slow symptom progression, we administered prednisone and azathioprine, achieving good results. Patients with symptoms of AAG may test negative for anti–ACh receptor antibodies and mainly present symptoms of sympathetic dysfunction, with no pupillary symptoms, and respond better to corticosteroids.9
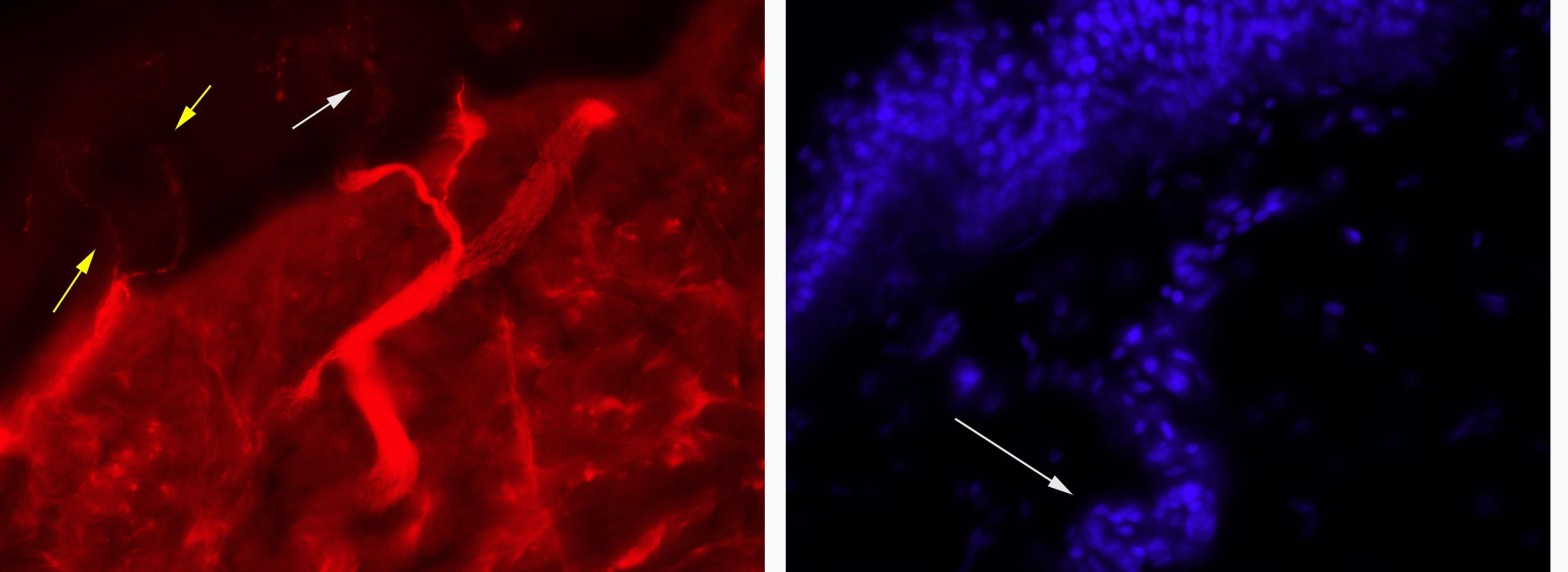
In this case, a morphological study revealed preserved innervation of the sweat glands, which supports the hypothesis of impaired transmission without associated neuronal or axonal injury (Fig. 1). However, prolonged ganglionic transmission blockade may have caused small fibre loss.10 Slowly progressive AAG may initially be misdiagnosed as PAF11; immunosuppresive therapy helps identify patients with AAG, who will respond to treatment, unlike those with PAF, a neurodegenerative disease.
: normal density of epidermal nerve fibres (10.1 fibres per mm, that is 0.9 standard deviations below the mean for the patient’s age and sex). Right (DAPI fluorescent stain): the arrow points to a normally innervated sebaceous gland.")
We would like to thank Dr Steven Vernino of University of Texas Southwestern Medical Center for his advice and for the antibody study.
Please cite this article as: Idiáquez Cabezas J, Riquelme Alcázar J, Calvo Bascuñán M, Casar Leturia JC. Ganglionopatía autonómica autoinmune crónica. Una causa rara de disautonomía. Neurología. 2021;36:388–390.
