




Fragile X syndrome, the most common inherited cause of intellectual disability, is associated with a broad spectrum of disorders across different generations of a single family. This study reviews the clinical manifestations of fragile X-associated disorders as well as the spectrum of mutations of the fragile X mental retardation 1 gene (FMR1) and the neurobiology of the fragile X mental retardation protein (FMRP), and also provides an overview of the potential therapeutic targets and genetic counselling.
DevelopmentThis disorder is caused by expansion of the CGG repeat (>200 repeats) in the 5 prime untranslated region of FMR1, resulting in a deficit or absence of FMRP. FMRP is an RNA-binding protein that regulates the translation of several genes that are important in synaptic plasticity and dendritic maturation. It is believed that CGG repeat expansions in the premutation range (55–200 repeats) elicit an increase in mRNA levels of FMR1, which may cause neuronal toxicity. These changes manifest clinically as developmental problems such as autism and learning disabilities as well as neurodegenerative diseases including fragile X-associated tremor/ataxia syndrome (FXTAS).
ConclusionsAdvances in identifying the molecular basis of fragile X syndrome may help us understand the causes of neuropsychiatric disorders, and they will probably contribute to development of new and specific treatments.
El síndrome X frágil (SXF) es la causa más frecuente de discapacidad intelectual hereditaria y se asocia a un amplio espectro de enfermedades en las distintas generaciones de una misma familia. En este trabajo se revisan las manifestaciones clínicas de los trastornos asociados al X frágil y el espectro de mutaciones en el gen 1 del retraso mental del X frágil (FMR1), la neurobiología de la proteína del retardo mental X frágil (FMRP) y una visión general de los potenciales blancos terapéuticos y el asesoramiento genético.
DesarrolloEsta enfermedad es causada por una amplificación de las repeticiones CGG (>200 repeticiones) en la región 5’ no traducida del gen FMR1, que lleva al déficit o ausencia de la proteína FMRP. La FMRP es una proteína de unión al ARN que regula la traducción de varios genes que son importantes en la plasticidad sináptica y la maduración dendrítica. Se cree que expansiones de las repeticiones CGG en el rango de premutación (55-200 repeticiones) generan un aumento en los niveles de mRNA de FMR1, lo que produciría toxicidad neuronal. Esto se manifiesta en problemas del desarrollo tales como autismo y problemas de aprendizaje, así como en patologías neurodegenerativas como el síndrome de temblor/ataxia asociado al X frágil (FXTAS).
ConclusionesLos avances en la identificación de las bases moleculares del SXF pueden servir como modelo para comprender las causas de las enfermedades neuropsiquiátricas y probablemente conducirán al desarrollo de tratamientos cada vez más específicos.
Around 1% to 3% of the world's population has intellectual disability (ID).1 Fragile X syndrome (FXS) is the most frequent cause of hereditary ID in men2 and the main monogenic disorder associated with autism.3 FXS affects one in 4000 men and one in 8000 women2; however, it may be more frequent if we consider mild ID and behaviour disorders.4 Nearly half of the cases of X-linked ID correspond to FXS.5
FXS has an X-linked dominant inheritance pattern and incomplete penetrance. It belongs to a group of disorders associated with mutations in the fragile X mental retardation 1 gene (FMR1), called fragile X-associated disorders (FXAD), which include fragile X-associated tremor/ataxia syndrome (FXTAS) and fragile X-associated primary ovarian insufficiency (FXPOI).6
FXS is clinically characterised by ID and such physical features as large protruding ears, hyperextensible joints, and flat feet.7 Men usually display strabismus, elongated face, prominent jaw, pectus excavatum, mitral valve prolapse, and macroorchidism after the age of 8.8,9 However, around 30% of the patients do not present either classic phenotypic traits or a family history of ID; rather, they display symptoms resembling language delay or attention-deficit/hyperactivity disorder (ADHD), which delays and hinders diagnosis.7
Symptoms of cognitive impairment appear early and are accompanied by psychomotor retardation,10 repetitive movements, unusual postures, poor visual contact, and social isolation.11 Cognitive impairment, language delays, and adjustment disorders are more severe in the 30% of children with autistic features.12 In some cases, psychomotor retardation may be mild; some children may even display normal psychomotor development initially, and psychomotor retardation may manifest as a learning disorder at a later stage.13 Around 85% of men and 25% to 30% of women with FXS have an IQ below 70; more women than men display average or borderline intellectual functioning.14 Most patients develop oral language and acquire general knowledge, and they are able to perform daily living activities.15,16
Some 13% to 18% of men and 4% of women with FXS present epilepsy in the form of generalised or partial complex seizures.17,18 Epilepsy is 3 times more frequent in patients with FXS with autistic features.17
Patients with FXS also display emotional disorders, including anxiety and hyperactivity; they may also show irritability, inflexibility, and aggressiveness.19,20 Emotional disorders, especially depression, anxiety, and shyness, are more frequent in women.21
Some patients have a similar phenotype to that of Prader-Willi syndrome: traits include ID, obesity, and delayed puberty, which may be associated with autism; patients with FXS do not have hypogonadism.22
Mutations in the fragile X mental retardation 1 gene and associated disordersIn more than 98% of the cases, FXS is caused by a full mutation (FM) due to the expansion of the CGG triplet (>200 repeats) in the 5′ untranslated region of the FMR1 gene, at the FRAXA locus on Xq27.3.23
Healthy individuals have between 4 and 45 CGG repeats in this region, which keeps it stable during DNA replication. Expansions containing more than 50 repeats make the region less stable, and may result in an FM in the following generations.24
The FM causes hypermethylation of the cytosine-phosphate-guanine (CpG) island located in the promoter region of the FMR1 gene, leading to conformational changes in chromatin, which becomes more compact. Chromatin condensation leads to complete or nearly complete inhibition of gene transcription, low or null levels of messenger ribonucleic acid (mRNA), and a significant decrease in or complete absence of the fragile X mental retardation protein (FMRP).25,26 However, FMR1 gene silencing has recently been found to be caused by the expanded mRNA of the gene via hybridisation between the complementary portions of CGG repeats, forming a RNA–DNA complex and preventing gene expression.27
Although less frequently, FXS may also be caused by a point mutation, a deletion of the FMR1 gene or its promoter,28 or short CGG repeat tracts (premutation [PM]) which is able to cause low levels of FMRP and thus ID.29,30
The phenotypic differences observed between women with FM, as well as between men and women with FM, are explained by the different levels of FMRP expression and non-random inactivation of the mutant X chromosome. Women with preferential inactivation of the X chromosome have more marked ID and low levels of FMRP31,32; their physical and neuropsychiatric phenotype is similar to that in men, although less severe.
Expansion of the CGG triplet to between 55 and 200 repeats, called PM, is a condition associated with instability during replication, and especially in female gametogenesis. In the general population, it has an incidence of one in 113 to 259 women and one in 260 to 813 men.2 PM does not affect expression of the FMRP substantially; however, expansions of more than 100 CGG repeats frequently yield an FM and FXS in the next generation.29
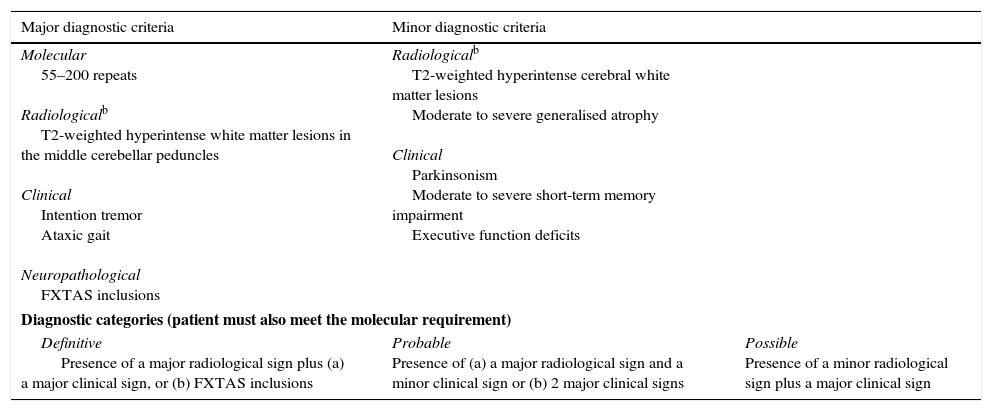
PM carriers, especially men, show mild cognitive impairment and behaviour disorders.33 Clinical impairment is associated with toxicity caused by high levels of mRNA of the FMR1 gene.8,34 PM is also associated with FXPOI (20% of female carriers), which manifests as cessation of menstrual periods before reaching the age of 40, and FXTAS in men (40%) and women (8%–16%) in adulthood.35,36 The diagnostic criteria of FXRAS are well established; although they are currently being revised, they are very useful for establishing a diagnosis of definitive, probable, or possible FXTAS (Table 1).37 Since 2% to 4% of men with late-onset cerebellar ataxia (starting after 50) may be carriers of a PM, we recommend studying the number of CGG repetitions in FMR1 in all patients older than 50 who display these symptoms.38,39 Male and female carriers of the PM are more frequently affected by autoimmune disorders, including hypothyroidism and fibromyalgia.37
Diagnostic criteria and diagnostic categories for FXTAS.a
| Major diagnostic criteria | Minor diagnostic criteria | |
|---|---|---|
| Molecular 55–200 repeats Radiologicalb T2-weighted hyperintense white matter lesions in the middle cerebellar peduncles Clinical Intention tremor Ataxic gait Neuropathological FXTAS inclusions | Radiologicalb T2-weighted hyperintense cerebral white matter lesions Moderate to severe generalised atrophy Clinical Parkinsonism Moderate to severe short-term memory impairment Executive function deficits | |
| Diagnostic categories (patient must also meet the molecular requirement) | ||
| Definitive Presence of a major radiological sign plus (a) a major clinical sign, or (b) FXTAS inclusions | Probable Presence of (a) a major radiological sign and a minor clinical sign or (b) 2 major clinical signs | Possible Presence of a minor radiological sign plus a major clinical sign |
Expansion of CGG repeats is mitotically unstable and favours somatic heterogeneity and presence of mosaic mutations. Mosaic carriers have FM alleles and PM alleles (12% of the patients with FXS).5,30 Patients may also display mosaic methylation patterns; this term refers to the co-presence of alleles with hypermethylated FM and demethylated alleles expanded to the PM and/or FM ranges (6% of the patients). Mosaic mutation carriers display varying levels of FMRP expression and a higher IQ.14,30
It is also important to highlight that CGG repetitions may be interrupted by AGG triplets, and the quantity and position of these triplets are important for replicative stability and therefore affect the probability of expansion in the next generation. In fact, the risk of expansion to an FM in a child of a PM carrier mother decreases by 60% when the mother presents 2 AGG interruptions within a total repeat length of 70 to 80 CGG repeats, compared to mothers with no interruptions within the same range.40
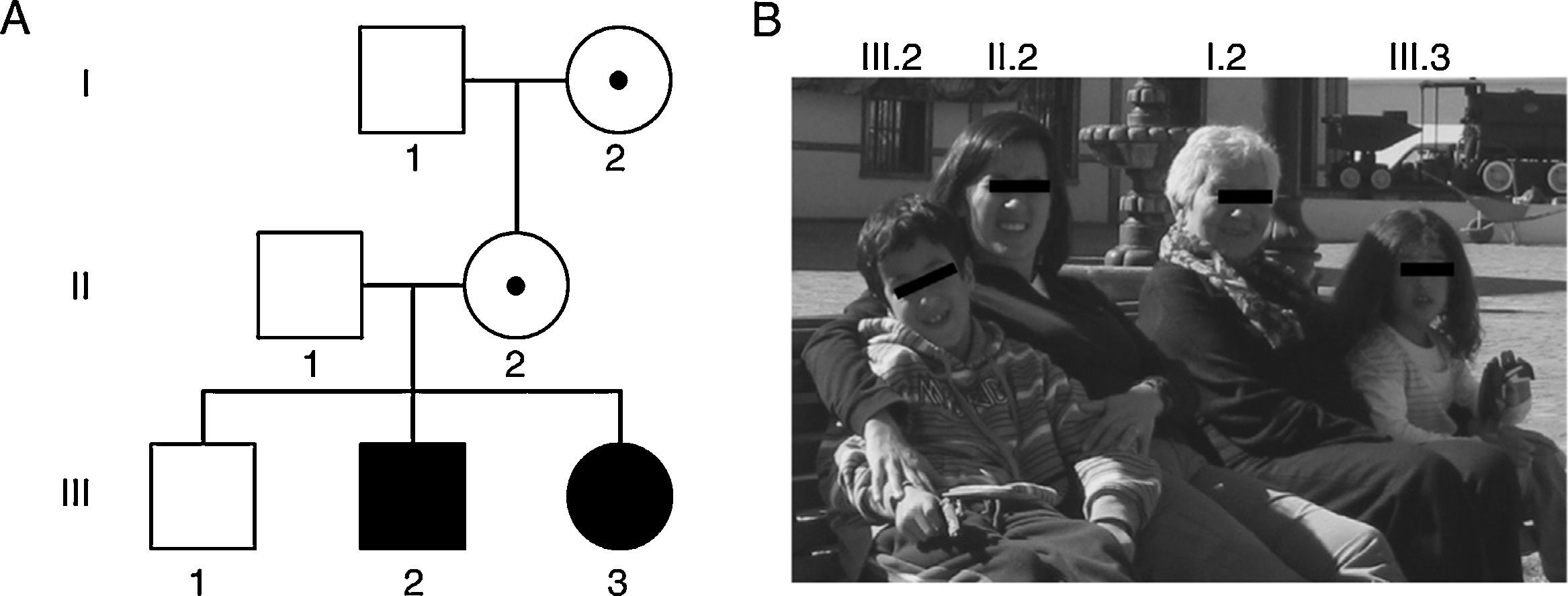
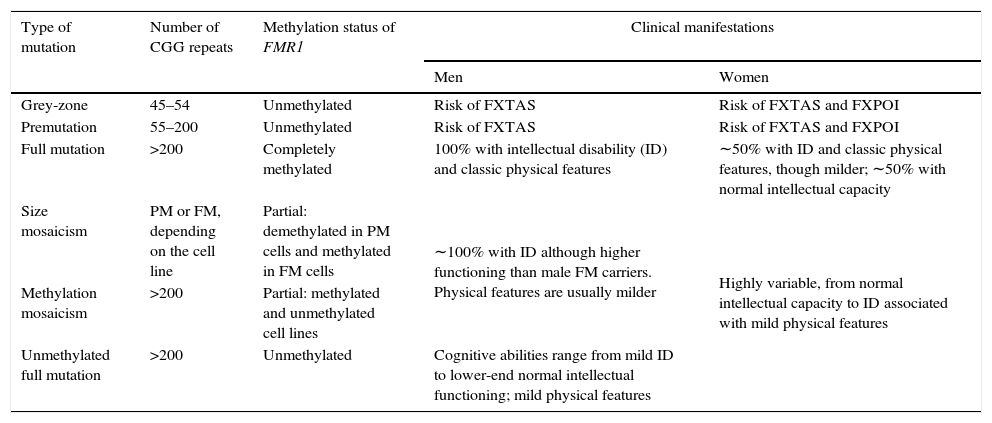
Lastly, expansions ranging from 45 to 54 CGG repeats are intermediate (also called ‘grey zone’) and combine normal and PM alleles. Grey-zone expansions are not clearly associated with a specific phenotype. They may however expand to an FM after 2 generations; carriers would have a slightly increased risk of developing FXTAS and/or FXPOI.37,41 Thus, the wide spectrum of disorders and symptoms associated with the mutations of the FMR1 gene lead families to display different genotypic and phenotypic manifestations in successive generations (Fig. 1 and Table 2).42,43
 Members I.2 and II.2 had PMs of 83 and 96 CGG triplets and manifested premature ovarian insufficiency at the ages of 35 and 36, respectively. Members III.2 and III.3 had FMs of 370 and 570 CGG triplets and were diagnosed with FXS at the ages of 5 and 1, respectively. (B) Photo of some members of the family indicating their positions in the genealogy.")
Family photo and genealogy showing the spectrum of fragile X-associated disorders. (A) Members I.2 and II.2 had PMs of 83 and 96 CGG triplets and manifested premature ovarian insufficiency at the ages of 35 and 36, respectively. Members III.2 and III.3 had FMs of 370 and 570 CGG triplets and were diagnosed with FXS at the ages of 5 and 1, respectively. (B) Photo of some members of the family indicating their positions in the genealogy.
Types of FMR1 mutations and associated clinical manifestations.a
| Type of mutation | Number of CGG repeats | Methylation status of FMR1 | Clinical manifestations | |
|---|---|---|---|---|
| Men | Women | |||
| Grey-zone | 45–54 | Unmethylated | Risk of FXTAS | Risk of FXTAS and FXPOI |
| Premutation | 55–200 | Unmethylated | Risk of FXTAS | Risk of FXTAS and FXPOI |
| Full mutation | >200 | Completely methylated | 100% with intellectual disability (ID) and classic physical features | ∼50% with ID and classic physical features, though milder; ∼50% with normal intellectual capacity |
| Size mosaicism | PM or FM, depending on the cell line | Partial: demethylated in PM cells and methylated in FM cells | ∼100% with ID although higher functioning than male FM carriers. Physical features are usually milder | Highly variable, from normal intellectual capacity to ID associated with mild physical features |
| Methylation mosaicism | >200 | Partial: methylated and unmethylated cell lines | ||
| Unmethylated full mutation | >200 | Unmethylated | Cognitive abilities range from mild ID to lower-end normal intellectual functioning; mild physical features | |
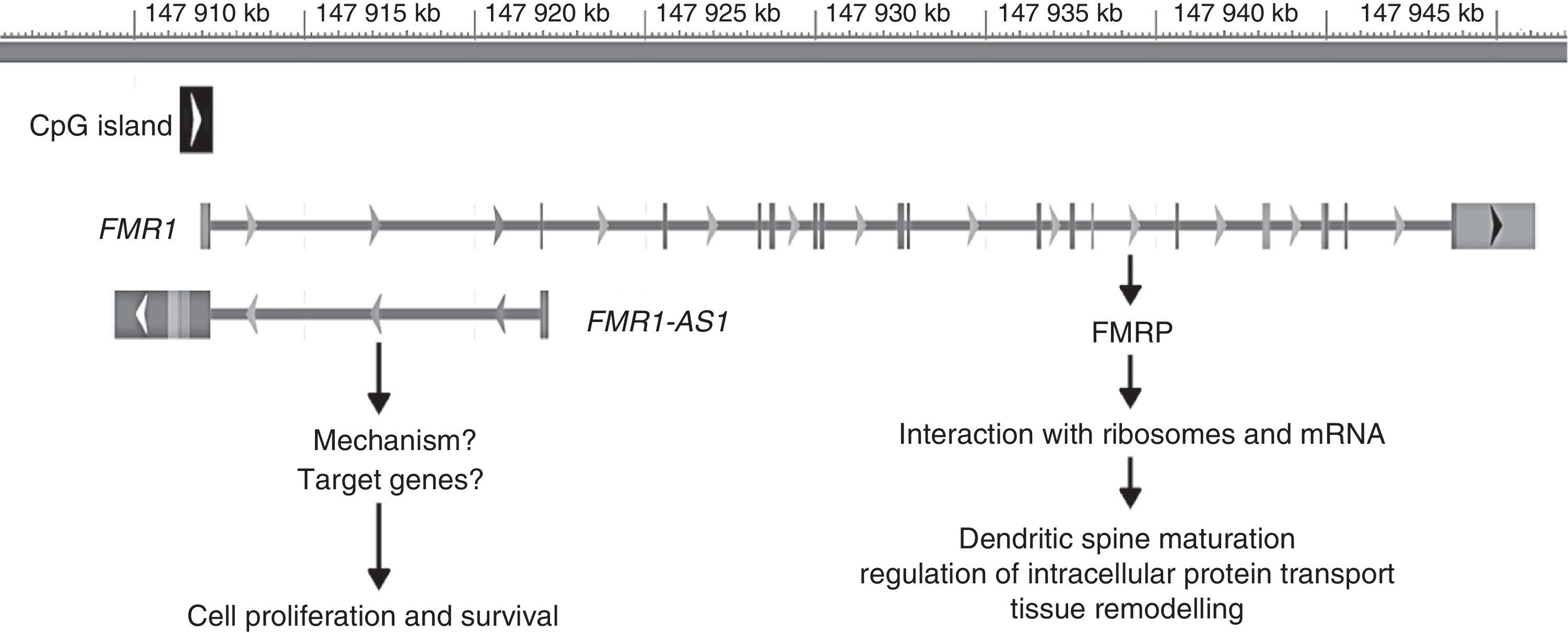
The FMR1 gene has recently been found to act via 2 mechanisms: the best known of these is associated with its protein product, FMRP, whereas the second mechanism is linked to another gene product, long non-coding RNA (lncRNA) FMR4 or FMR1-AS1. LncRNA are genes coding for RNA that participate in transcriptional regulation of other genes, which can be positive or negative.44,45 The promoter of FMR1 codes for the FMR4 gene in antisense orientation; this gene overlaps the CGG repeat region and the CpG island. As occurs with FMR1, FMR4 is silenced in patients with FM and upregulated in PM carriers (Fig. 2).45
![Genomic location and role of FMR1 and FMR1-AS1. The grey bar represents the genomic position (in kilobases [kb]) of both genes on the X chromosome from the P-terminal end. The black square represents the CpG island involved in silencing both genes in patients with FM. The arrows placed next to the CpG island and each gene represent the direction of transcription. The vertical lines in FMR1 represent the exons of this gene.](https://static.elsevier.es/multimedia/21735808/0000003200000004/v1_201704190057/S2173580816301249/v1_201704190057/en/main.assets/gr2.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNcnxJVYM2BEAEuZ5XZLhi9i6WSMfelRHtJaOSE9bGYOuZpAEskOvZ38NrvZTgRjNkoTsjvtRBjaFr2r8U2wpzf0a1sLH0JC6Isv2/PvlVoy+U8gg07q+nY2nw20IOqVvmKaM/IAO2lrVtbOMhcxwwhyIvsMMxp+M6cKlu25srlqLMA4fX4gRFUy3VdBxzYTUltR7E4eYPtGNDXlRnZK1XdmqBrTuY8l2XzU4Gl0vBQ+2VpedTfOFqdqUnLXRxObiQQ= "Genomic location and role of FMR1 and FMR1-AS1. The grey bar represents the genomic position (in kilobases [kb]) of both genes on the X chromosome from the P-terminal end. The black square represents the CpG island involved in silencing both genes in patients with FM. The arrows placed next to the CpG island and each gene represent the direction of transcription. The vertical lines in FMR1 represent the exons of this gene.")
Genomic location and role of FMR1 and FMR1-AS1. The grey bar represents the genomic position (in kilobases [kb]) of both genes on the X chromosome from the P-terminal end. The black square represents the CpG island involved in silencing both genes in patients with FM. The arrows placed next to the CpG island and each gene represent the direction of transcription. The vertical lines in FMR1 represent the exons of this gene.
FMRP, with a maximum of 631 aminoacids, has 2 RNA-binding motifs, a nuclear localisation signal, a nuclear export signal, and 2 protein–protein interaction domains.46 FMRP expression levels vary depending on the tissue and the type of cell within a tissue. In humans, for example, this protein is widely expressed in the epithelium and the central nervous system. Within the central nervous system, FMRP is expressed in the brainstem, the structures arising from the forebrain, and the cerebellum; expression is more marked in neurons than in glial cells. Lastly, within neurons, FMRP is more abundant in the soma, proximal dendrites, and synapses.47
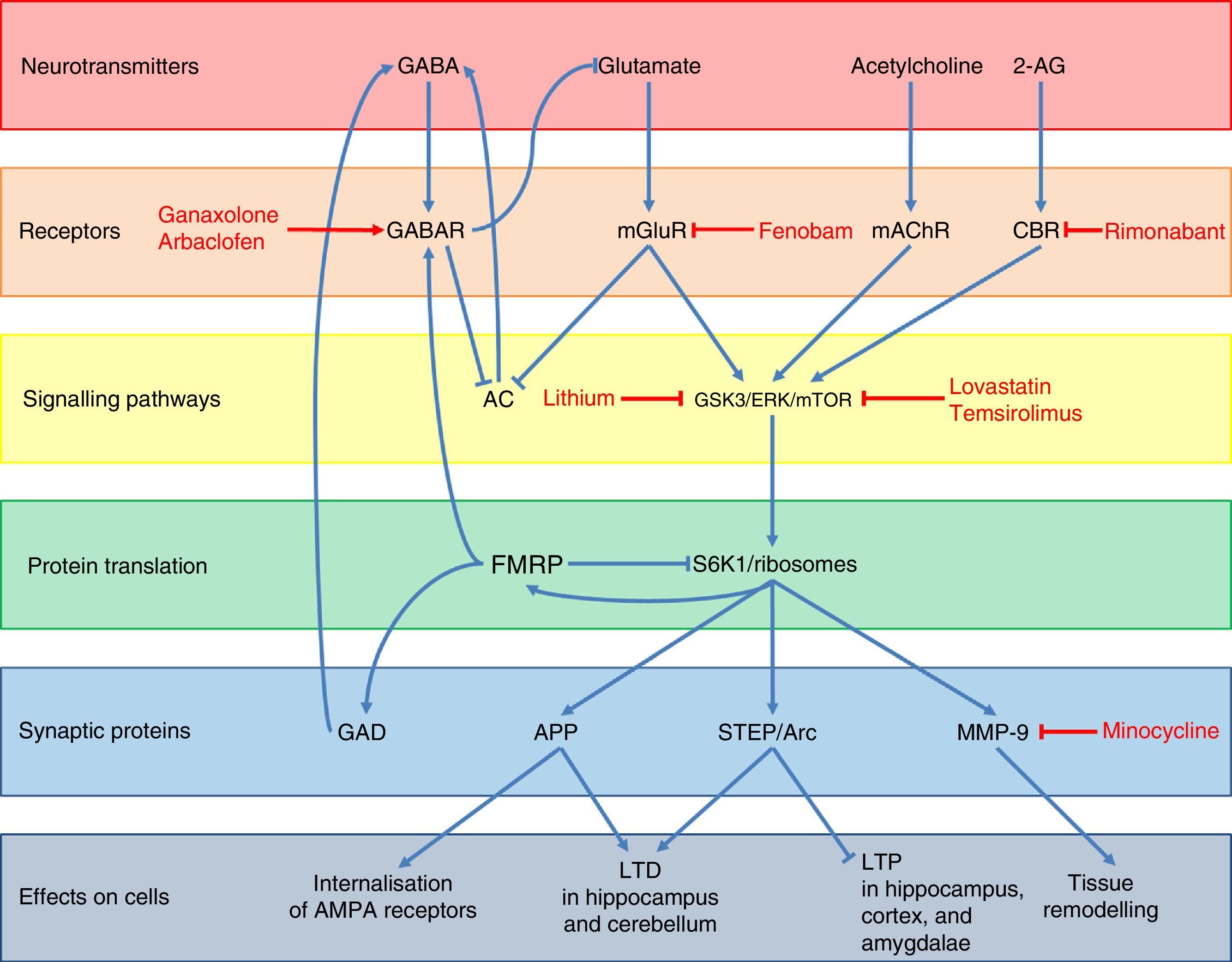
Regarding FMRP function, several pathways used by this protein to regulate neuronal synapsis and the expression of other genes have been described (Fig. 3).48 Under normal conditions, FMRP binds to a wide range of mRNA targets,49 interacts with the 80S ribosome,50 and decreases ribosomal protein S6 kinase 1 (S6K1) activity.49 This prevents the translation of different synaptic proteins including the amyloid precursor protein (APP), the striatal-enriched protein tyrosine phosphatase (STEP), the activity-regulated cytoskeleton-associated protein (Arc), and matrix metalloproteinase 9 (MMP-9).48,51,52 These proteins promote internalisation of AMPA receptors, long-term depression (LTD) in the hippocampus and cerebellum, and tissue remodelling at the synaptic level, while decreasing long-term potentiation (LTP) in the hippocampus, cortex, and amygdalae.48,49,51 In addition, FMRP promotes the expression of GABAA receptors and glutamic acid decarboxylase (GAD), the rate-limiting enzyme for GABA synthesis.48,53 This means that FMRP participates in dendritic spine maturation, synaptogenesis,54 and dendritic transport and transport regulation of proteins.55 Likewise, FMRP acts as a translational repressor at synapses, regulating mRNA levels of proteins involved in synaptic structure and function,56 and it plays a crucial role in the formation of regulated mRNA patterns at the subcellular level during development.57

Diagram of the neurobiology of FMRP and the different therapeutic targets and drugs used for FXS according to the cellular components with which that protein interacts. Ganaxolone and arbaclofen are GABAA and GABAB receptor agonists, respectively; fenobam is an mGluR5 antagonist; and rimonabant is a cannabinoid CB1 receptor antagonist. Targets in signal transduction pathways are GSK3, ERK, and mTOR, which are inhibited by lithium, lovastatin, and temsirolimus, respectively. Lastly, minocycline inhibits the matrix metalloproteinase MMP-9.
A lack of FMRP results in a wide range of alterations in different neurotransmitter systems, the most important and most studied alteration being dysregulation of glutamatergic pathways, especially via metabotropic glutamate receptors (mGluR) and their downstream proteins. A loss in the regulatory function of FMRP results in overexpression of APP, STEP, Arc, and MMP-9; these are downstream effectors of the mGluR pathway (Fig. 3).48,51 Glutamatergic pathway overactivation is also due to lack of counterregulation in the GABA pathway due to GABAA receptor underexpression and decreased GABA synthesis.53 This system also inhibits glutamate release: a lack of FMRP indirectly results in increased mGluR activation.48 In addition, mGluR also lead to decreased activity of adenylyl cyclase (AC)58 and the subsequent decrease in GABA release. Lastly, the endocannabinoid and acetylcholine systems are also involved in increased activation of the glutamatergic pathway as they interact with mTOR and ERK proteins; these are downstream mGluR transduction proteins which are activated by cannabinoid receptors (CBR) and muscarinic acetylcholine receptors (mAChR) (Fig. 3).48,59 The above yields weak synaptic connections60–63 and greater susceptibility to seizures.64
In the PM range, FMR1 is transcribed efficiently and FMRP expression is nearly normal, whereas mRNA translation is poor and compensated with higher levels of mRNA (a 5- to 8-fold increase).14,34 Thus, ubiquitin-positive inclusions accumulate in the nuclei of neurons and glial cells65 containing expanded FMR1 mRNA and sequestered RNA-binding proteins, which affects the function of these proteins and causes neurodegeneration.66 In humans, the number and size of inclusions increase with age and disease progression.67 Additionally, mitochondrial dysfunction has been found in fibroblasts and brain samples from PM carriers, which results in increased oxidative stress and lower expression of mitochondrial proteins.68
The role of FMR1-AS1 in the pathogenesis of FXS is not well understood. Khalil et al.69 showed that partial silencing of FMR4 resulted in alterations in the cell cycle and increased apoptosis but had no impact on FMR1 expression and vice versa, which suggests that FMR1 has an independent mechanism. Many questions regarding the function of FMR1 and FMR1-AS1 must still be answered (Fig. 2). Nevertheless, we may conclude that FXS arises due to lack of expression of both genes, whereas disorders associated with PM are due to FMR1 and FMR1-AS1 mRNA overexpression, which leads to cell degeneration.
Confirmation of diagnosisTests for detecting FMR1 mutations are sensitive and specific for both patients manifesting the disease and asymptomatic carriers of the mutation. These tests are conducted on DNA extracted from a blood sample; the gene is analysed directly to determine the number of CGG repeats and methylation status of the locus.70,71
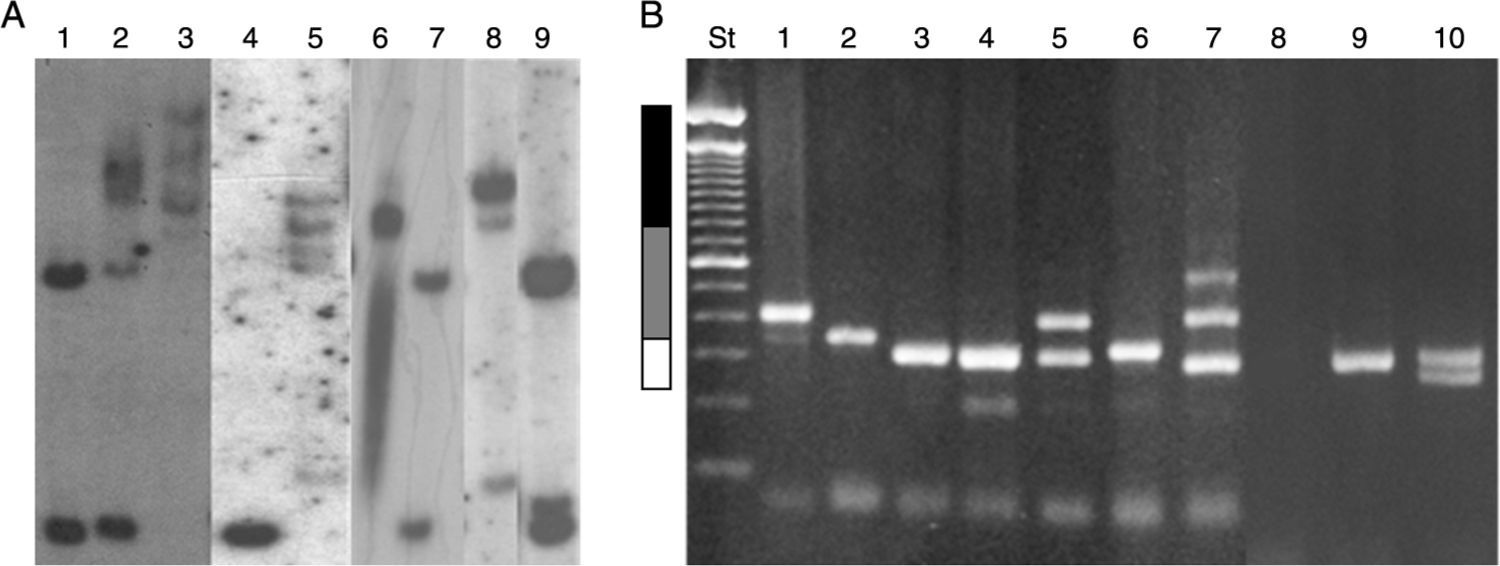
Numerous molecular methods for analysing mutations at the FRAXA locus are currently in use: (1) Techniques based on PCR (polymerase chain reaction) are usually fast, simple, and inexpensive, especially when used to test men only. These techniques can also determine the exact size of the alleles in the normal, grey-zone, PM, and FM ranges, even in women.72,73 However, these results must be confirmed by Southern blot analysis.70 (2) Southern blot provides a direct analysis of the FRAXA locus and the FMR1 gene and determines the size of the expansion and the gene's methylation status; these data are used to determine the level of FMRP expression. These techniques are currently available in Chile, although costs are not covered by health insurance providers (Fig. 4).71,74 (3) Real-time quantitative PCR-based techniques and methylation specific-quantitative melt analysis (MS-QMA) have recently been developed. These techniques determine the methylation status of other CpG loci within FMR1, and their results are correlated with the level of FMRP expression and the patients’ cognitive status.75
 Southern blot results. Lanes 1 and 7: female non-carrier; lane 2: female FM carrier; lane 9: female PM carrier; lane 4: male non-carrier; lane 3: male FM carrier; lanes 5 and 8: male mosaic (PM/FM) carriers; lane 6: partially demethylated male FM carrier. (B) Diagnostic results from PCR. Lanes 1 and 2: male PM carriers; lanes 3, 6, and 9: male non-carriers; lane 4: female non-carrier (homozygosity confirmed by Southern blot); lane 5: female PM carrier; lane 7: female mosaic (PM/FM) carrier; lane 8: male FM carrier; lane 10: female non-carrier (heterozygous). St: molecular weight standard of 100bp (Invitrogen). White bar: normal range (< 55 CGG repeats). Grey bar: PM range (55–200 CGG repeats). Black bar: FM range (> 200 CGG repeats).")
Molecular results in the diagnosis of FMR1-related disorders. (A) Southern blot results. Lanes 1 and 7: female non-carrier; lane 2: female FM carrier; lane 9: female PM carrier; lane 4: male non-carrier; lane 3: male FM carrier; lanes 5 and 8: male mosaic (PM/FM) carriers; lane 6: partially demethylated male FM carrier. (B) Diagnostic results from PCR. Lanes 1 and 2: male PM carriers; lanes 3, 6, and 9: male non-carriers; lane 4: female non-carrier (homozygosity confirmed by Southern blot); lane 5: female PM carrier; lane 7: female mosaic (PM/FM) carrier; lane 8: male FM carrier; lane 10: female non-carrier (heterozygous). St: molecular weight standard of 100bp (Invitrogen). White bar: normal range (< 55 CGG repeats). Grey bar: PM range (55–200 CGG repeats). Black bar: FM range (> 200 CGG repeats).
A study in animal models of FXS has shown that exposure to an enriched environment may alleviate behavioural disorders; in this study, changes were assessed using nesting material, physical exercise, and a wide range of plastic toys of different colours and textures.76 Patients with FXS have been found to benefit from occupational and speech therapy.77 As a general rule, implementing specific techniques in education centres and the family setting leads to significant improvements in behaviour and autistic symptoms in children with FXS.31
Pharmacological treatmentThere are 2 types of pharmacological treatment for FXS: treatment for neuropsychiatric symptoms and treatment focused on the pathophysiology underlying the disease.
Treatment of clinical manifestationsThis type of treatment focuses on symptoms and disorders associated with FXS, including ADHD, anxiety, behaviour disorders, and seizures. The most widely used drugs are psychostimulants (for attention deficit and hyperactivity), serotonin reuptake inhibitors (aggressiveness associated with anxiety), and atypical antipsychotics (irritability).78,79 Such α-adrenergic receptor agonists as clonidine and guanfacine are preferred for treating ADHD in children younger than 5.79l-Acetylcarnitine has been found to improve ADHD symptoms in controlled pilot studies.80 Trials of sertraline in small groups have shown decreased anxiety,81 and a study of aripiprazole including 15 patients reported significant improvements in irritability.78 However, randomised double-blind trials with larger numbers of patients should be conducted to validate these treatments.
For patients with seizures, carbamazepine and valproic acid are the current treatments of choice since they are able to control epilepsy and cause few adverse effects.17,82 Both lamotrigine and levetiracetam have proven to be effective additions to anticonvulsant treatment in refractory cases, while causing minimal cognitive adverse effects.79 Phenytoin, phenobarbital, and gabapentin are usually avoided since they either cause adverse effects or exacerbate behavioural disorders.79
Experimental treatments based on the pathophysiology of fragile X syndromeAdvances in our knowledge of the neurobiology of FXS have led to the development of a range of drugs which act on the neurotransmitter pathways involved in this disease. As with the pathophysiological mechanisms described in Fig. 3, experimental pharmacological treatments can be classified into those acting on the receptors of the neurotransmitters involved in FXS, those acting on intracellular signalling proteins downstream of neurotransmitters, or those acting on synaptic effector proteins.
The first group includes GABA receptor agonists and different glutamatergic agonists, which compensate overactivation of the glutamatergic pathway caused by FMRP deficits (Fig. 3).48 This explains how GABAA receptor agonists such as ganaxolon control seizures and anxiety in patients with FXS,83 although they can also cause drowsiness.84,85 Baclofen, a GABAB receptor agonist, has been shown to be effective for treating hyperactivity and seizures in FMR1 knockout mice.86 Arbaclofen, an isomer of baclofen that is significantly more potent than regular baclofen as a GABA agent, has been shown to decrease irritability and improve social interaction in humans,87,88 although further studies should evaluate its toxicity and long-term effects. According to several studies, fenobam, an antagonist of metabotropic glutamate 5 receptor (mGluR5), effectively improves some behavioural symptoms and reduces dendritic anomalies in the hippocampus of FMR1 knockout mice.48 A preliminary study including 12 adult patients with FXS treated with a single dose of fenobam showed significant improvements in social interaction and hyperactivity; however, some patients experienced mild sedation and greater anxiety.89 Several clinical trials of other mGluR5 antagonists are currently underway.90 Regarding the endocannabinoid system, rimonabant is being tested on animals; this cannabinoid CB1 receptor antagonist prevents interaction between CB1 receptor and its endogenous ligand, 2-arachidonoylglycerol (2-AG).48 Short-term treatment with rimonabant in FMR1 knockout mice has been found to completely or partially normalise object-recognition memory and susceptibility to audiogenic seizures, whereas long-term treatment improved dendritic spine density and decreased mTOR signalling in the hippocampus.59
Several pharmacological treatments which inhibit the proteins involved in intracellular signalling downstream of mGluR are currently being developed (Fig. 3).48 Glycogen synthase kinase-3 (GSK-3) is overactive in FMR1 knockout mice.48 Lithium, a GSK-3 inhibitor, is usually well tolerated and has been shown to significantly improve behaviour, adaptive skills, and verbal memory in a pilot study including 15 young patients with FXS.91 Lovastatin and temsirolimus (ERK and mTOR inhibitors, respectively) reduced memory deficits, susceptibility to audiogenic seizures, and the synthesis of proteins involved in LTD in FMR1 knockout mice.48
Lastly, inhibitors of synaptic effector proteins are also being explored.48 The inhibitory activity of minocycline on MMP-9 has been found to significantly improve intellectual function and decrease anxiety in mice by promoting dendritic spine maturation in the hippocampus.92 In one preliminary study in humans, minocycline was well tolerated and decreased irritability, stereotypy, hyperactivity, and inappropriate speech, but controlled trials have yet to be performed.93
No systematic reviews on these pharmacological treatments have been conducted to date due to low patient numbers, short treatment duration, and the difficulties in comparing results from different assessment tools. Although current treatment strategies have an effect on symptoms, they do not improve the cognitive profile of these patients. As a general rule, combination therapy (pharmacological and non-pharmacological treatment) provides greater benefits.94
Treatment for fragile X-associated primary ovarian insufficiency and fragile X-associated tremor/ataxia syndromeTreatment for fragile X-associated primary ovarian insufficiencyWomen with PM and FXPOI may exhibit symptoms of menopause in addition to the psychological effect of premature loss of their reproductive ability. Even when no specific treatments are available, psychotherapy may be beneficial for patients with FXPOI.95 We should stress, however, that FXPOI does not rule out the possibility of pregnancy; the reproductive potential of women with this syndrome should therefore be evaluated by a gynaecologist. Likewise, due to these patients’ low serum levels of estradiol and its associated multisystemic consequences, the possibility of hormone replacement therapy should be evaluated by an endocrinologist.39
Treatment for fragile X-associated tremor/ataxia syndromePharmacological treatment of psychiatric disorders associated with PM is non-specific and includes conventional psychoactive drugs. Selective serotonin reuptake inhibitors are the most widely used medications for mood disorders.95
No specific treatments have been developed for FXTAS, although some drugs may partially improve symptoms. Propranolol and primidone are the most frequently used drugs for intention tremor. Some studies have reported improvements in patients treated with botulinum toxin, levetiracetam, clonazepam, clozapine, nadolol, and nimodipine.43 Physical therapy, amantadine, and buspirone have been shown to be beneficial for treating ataxia.96 Antipsychotics should be used with caution as they may aggravate movement disorders. Quetiapine is associated with a lower risk of extrapyramidal side effects.43,95
Although no formal recommendations have been established, PM carriers should undergo analyses of serum levels of thyroid stimulating hormone, free thyroxine, and triiodothyronine at least once a year to detect and treat hypothyroidism as early as possible.97
Evidence is insufficient to support the use of dementia drugs in patients with FXTAS. However, a recent study showed that administering memantine for a year significantly improved verbal memory in patients with FXTAS,98 laying the foundation for further clinical trials of this drug.
To date, there have been no experimental studies in which interfering RNA was able to counteract the high levels of mRNA in FMR1 in patients with PM. However, RNA intereference constitutes a potential therapeutic target based on experience with oligonucleotides in myotonic dystrophy type 1.37
Genetic counsellingWhen FXS is suspected, the family must be informed of the possible implications of conducting a genetic study, not only for the patient but also for other family members. Doctors must also stress the importance of referring the patient to a clinical geneticist.42 A geneticist will assist in family planning by compiling the family's medical history and making it available to them while providing timely information about the risks of bearing a mutation.42,99
The risk of transmitting the mutation to offspring depends on the bearer's sex and the number of repeats. Male PM carriers will transmit the mutation to all daughters but not to their sons. Female PM carriers have a 50% chance of transmitting the mutated allele in the PM or FM range to any of their offspring. The children of women with FM have a 50% risk of inheriting the FM. The sons of men with FM do not inherit the mutation whereas their daughters may inherit the FM or a PM due to contraction in the CGG repeat number (Table 3).99
Risk of transmitting the mutation to offspring.
| Parent | Type of mutation | Risk of having an affected son | Risk of having an affected daughter |
|---|---|---|---|
| Man | Premutation | 0% | 100% premutation |
| Woman | Premutation | 50% full mutation or premutationa | 50% full mutation or premutationa |
| Man | Full mutation | 0% | 100% full mutation |
| Woman | Full mutation | 50% full mutation | 50% full mutation |
All family members at risk of carrying PM or FM must be screened for neurological, emotional, and endocrine disorders. Families with mutation carriers may join a support group, especially those for parents of children with fragile X disorders, which are now active in many countries. It is also necessary to gather data about the number of AGG interruptions in PM female carriers in the Latin American population to determine how these interruptions have affected the probability of expansions in the offspring. This is recommended by the clinical guidelines for diagnosis and treatment of FMR1-associated diseases recently published in Spain and based on evidence from the population of the United States.40,100
The population with FXS has been studied from many different perspectives, including prenatal diagnosis, disease detection in populations with neurological disorders, and newborn screening.101 Although the subject is controversial, early diagnosis with newborn screening will permit early treatment, family counselling, and informed decision making.102 This may result in fewer new cases of FXS when carriers are diagnosed before a second child with the disease is born (Fig. 1).
ConclusionsFXS is associated with a wide range of clinical manifestations, from the classical phenotype of FM carriers to the neurological and psychiatric symptoms linked to the PM.37 Given this wide range of presentations and the high frequency of the disease, there is a high probability of coming across a patient with FXS at some point during our clinical careers. A molecular study of FMR1 must be considered in patients with psychomotor retardation, ID, autism, premature menopause, ataxic gait or intention tremor, parkinsonism, peripheral neuropathy, dementia, anxiety, or depression, especially when there is a family history of ID and/or autism.99,103
Psychiatric symptoms in PM carriers may be a primary manifestation of the PM and should therefore not be attributed solely to the stress generated by caring for a child with a disability (although this factor is always present).39
Molecular studies and genetic counselling (including extended genealogies) are essential for identifying family members with an FMR1 mutation and determining individual prognosis and risk of transmitting the disease to offspring.99
Advances in our understanding of the molecular basis of SXF may help clarify the aetiology of neuropsychiatric disorders and ascertain the mechanisms of neurological and psychiatric symptoms of other genetic diseases. This is likely to lead to increasingly specific treatments.48
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Pugin A, Faundes V, Santa María L, Curotto B, Aliaga S, Salas I, et al. Aspectos clínicos, moleculares y farmacológicos en los trastornos asociados a gen 1 del retraso mental del X frágil. Neurología. 2017;32:241–252.

Neurología (English Edition) sigue las recomendaciones para la preparación, presentación y publicación de trabajos académicos en revistas biomédicas