



Wernicke encephalopathy (WE) is the main neurological disorder caused by deficiency of the cofactor thiamine, which is crucial in energy metabolism; magnesium is also involved as a thiamine cofactor.1 WE can be alcoholic or non-alcoholic2 (caused by malnutrition or increased water-soluble vitamin loss, as in dialysis). Prevalence of WE lesions in autopsy studies has been reported to be 12.5% in alcoholics3 and 30% to 60% in alcohol-related fatalities.4,5 WE is a little-recognised and underdiagnosed condition. Although WE is more prevalent in men, women are more susceptible.6,7 Diagnosis is clinical and early treatment is fundamental in preventing coma and death.
We present the case of an 81-year-old woman (height 1.58m, weight 58kg, BMI 23.2), who was autonomous in the activities of daily living before symptom onset.
Her personal history included 10 years of schooling, no history of alcohol abuse, hypertension, hiatal hernia diagnosed 18 years previously, anti-reflux surgery 15 years previously, cholecystectomy, and acute biliary pancreatitis. She was being treated with pantoprazole, domperidone, ursodeoxycholic acid, candesartan, mexazolam, mirtazapine, and brotizolam.
Two to 3 weeks after an influenza episode, she developed anorexia, dehydration, mental confusion, altered sleep-wake cycle, and visual and gait impairment.
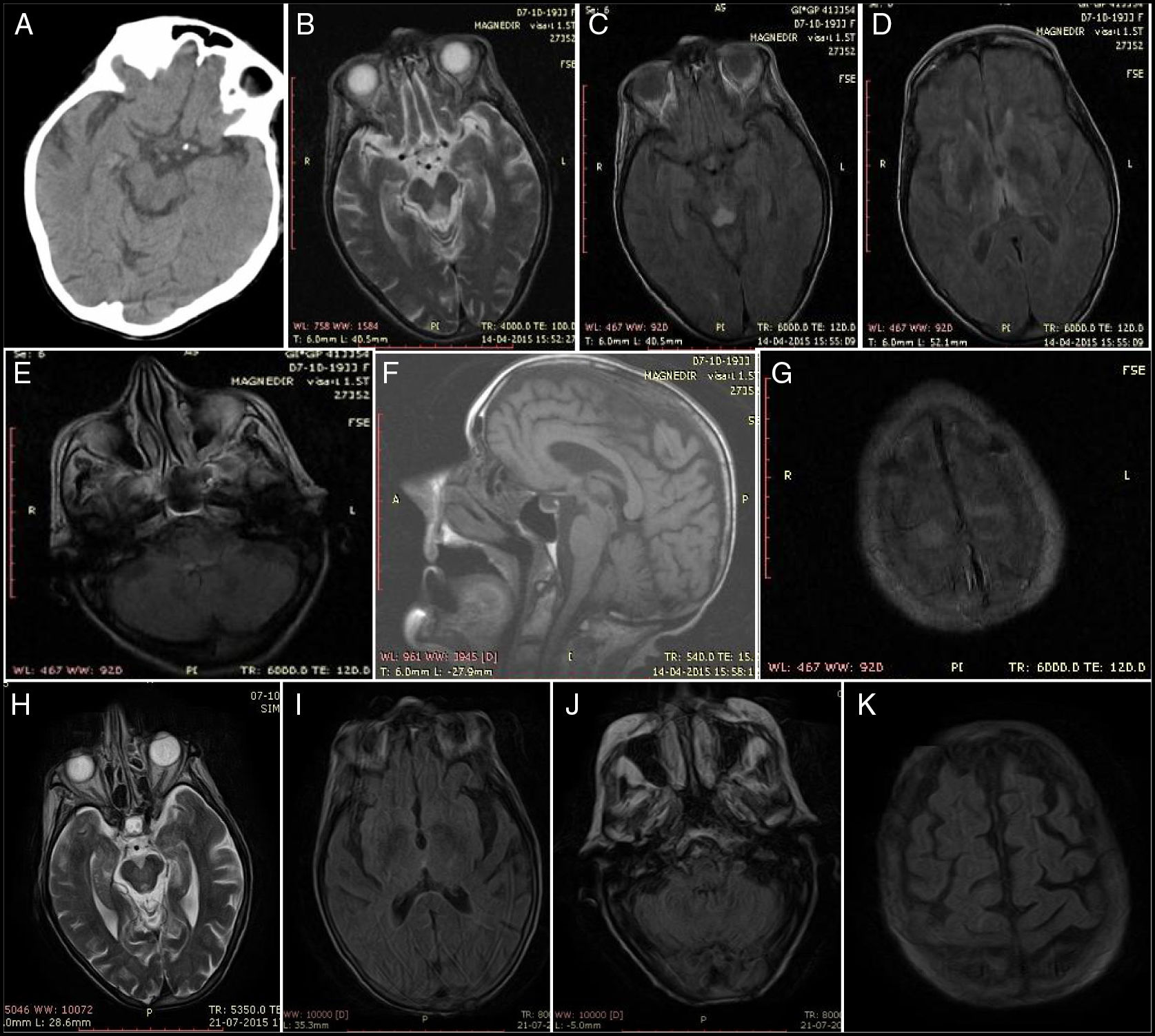
Physical examination revealed somnolence, disorientation in time but not space, incoherent speech, strabismus, persistent horizontal-rotary nystagmus, dysphagia for liquids, and hypotonia. The patient was haemodynamically stable, with normal results in the cardiopulmonary auscultation and sinus rhythm in the electrocardiography. Blood analysis revealed no anaemia or leukocytosis and a normal level of C-reactive protein. Urine benzodiazepine levels were twice the normal value. Head CT scan findings were normal but at 24hours she presented a suspected lacunar stroke of the right tectal plate (Fig. 1A), requiring examination with brain MRI. A transthoracic echocardiography did not detect cardioembolism, and lumbar puncture returned normal results.
 Head CT at 24hours: hypodensity in the right tectal plate. (B) Axial T2-weighted MRI sequence. (C) Axial FLAIR MRI sequence: lesion to the midbrain periaqueductal region. (D) Axial FLAIR MRI sequence: bilateral thalamic lesions. (E) Axial FLAIR MRI sequence: tectal lesion. (F) Sagittal T1-weighted MRI sequence: no alterations. (G) Axial FLAIR MRI sequence: lesion to the superior frontal cortex and pia mater. Findings at resolution: (H) Axial T2-weighted MRI sequence: regressing periaqueductal lesion. (I) Axial FLAIR MRI sequence: no thalamic lesion. (J) Axial FLAIR MRI sequence: no bulbar lesion. (K) Axial FLAIR MRI sequence: no lesion to the cortex or pia mater.")
Baseline findings: (A) Head CT at 24hours: hypodensity in the right tectal plate. (B) Axial T2-weighted MRI sequence. (C) Axial FLAIR MRI sequence: lesion to the midbrain periaqueductal region. (D) Axial FLAIR MRI sequence: bilateral thalamic lesions. (E) Axial FLAIR MRI sequence: tectal lesion. (F) Sagittal T1-weighted MRI sequence: no alterations. (G) Axial FLAIR MRI sequence: lesion to the superior frontal cortex and pia mater. Findings at resolution: (H) Axial T2-weighted MRI sequence: regressing periaqueductal lesion. (I) Axial FLAIR MRI sequence: no thalamic lesion. (J) Axial FLAIR MRI sequence: no bulbar lesion. (K) Axial FLAIR MRI sequence: no lesion to the cortex or pia mater.
She started treatment with thiamine at high doses (500mg IV every 8hours for 2 days, 500mg IV every 24hours for 5 days), then at 100mg IV every 8hours during the remaining days of hospitalisation, combined with a multivitamin solution (vitamin A, B, H [biotin], and F) and protein-calorie supplementation.
She was initially admitted to the stroke unit to rule out brainstem stroke. We observed a significant clinical improvement, with decreased nystagmus, improved verbal expression, and corrected sleep pattern. A brain MRI (Fig. 1B-G) performed at day 5 of admission revealed diffuse hyperintensity of the tectum, periaqueductal region, medial thalami, mammillary bodies, and structures adjacent to the diencephalon and cortical convexity with brain atrophy; these findings are indicative of WE.
At day 6 of admission, the patient was transferred to the neurology department. She was awake, with no spontaneous verbal response, strabismus (exotropia of the right eye), isochoric and reactive pupils (preserved photomotor and consensual reflexes), and mild horizontal-rotatory nystagmus. The patient presented no motor deficit and did not collaborate in the examination. Enteral feeding via nasogastric tube was continued; the patient's body weight was 39kg (BMI: 15.6).
The examination revealed a haemoglobin level of 7.8g/dL; haematocrit, 24% (normal range, 36-46); vitamin B1, 27ng/mL (28-85); vitamin B12, 158pg/mL (187-883); vitamin D, 17ng/mL (30-100); magnesium, 1.37mg/dL (1.6-2.6); sodium, 135mg/dL (136-145); proteins, 5.3g/dL (6.4-8.3), and albumin, 2.9g/dL (3.2-4.6). Results of the analysis of MCV and MCH (red blood cells), folic acid, ammonia, thyroid function, and calcium and phosphate metabolism were normal. Intrinsic factor antibody test and serological test for syphilis yielded negative results.
She received a transfusion of 1U of red blood cells; thiamine was maintained at 100mg IV every 8hours; pantoprazole was withdrawn and ranitidine started at 150mg at night. The patient also started treatment with oral vitamin B12 at 5mg/day, cholecalciferol 667IU/day, magnesium 10mL/12h, calcium carbonate 500mg/12h, and 0.9% saline solution.
During the first 2 weeks of progression, her speech improved and she was able to produce sentences; nystagmus manifested only at extreme lateral gaze. Ataxic gait was later identified and she started rehabilitation.
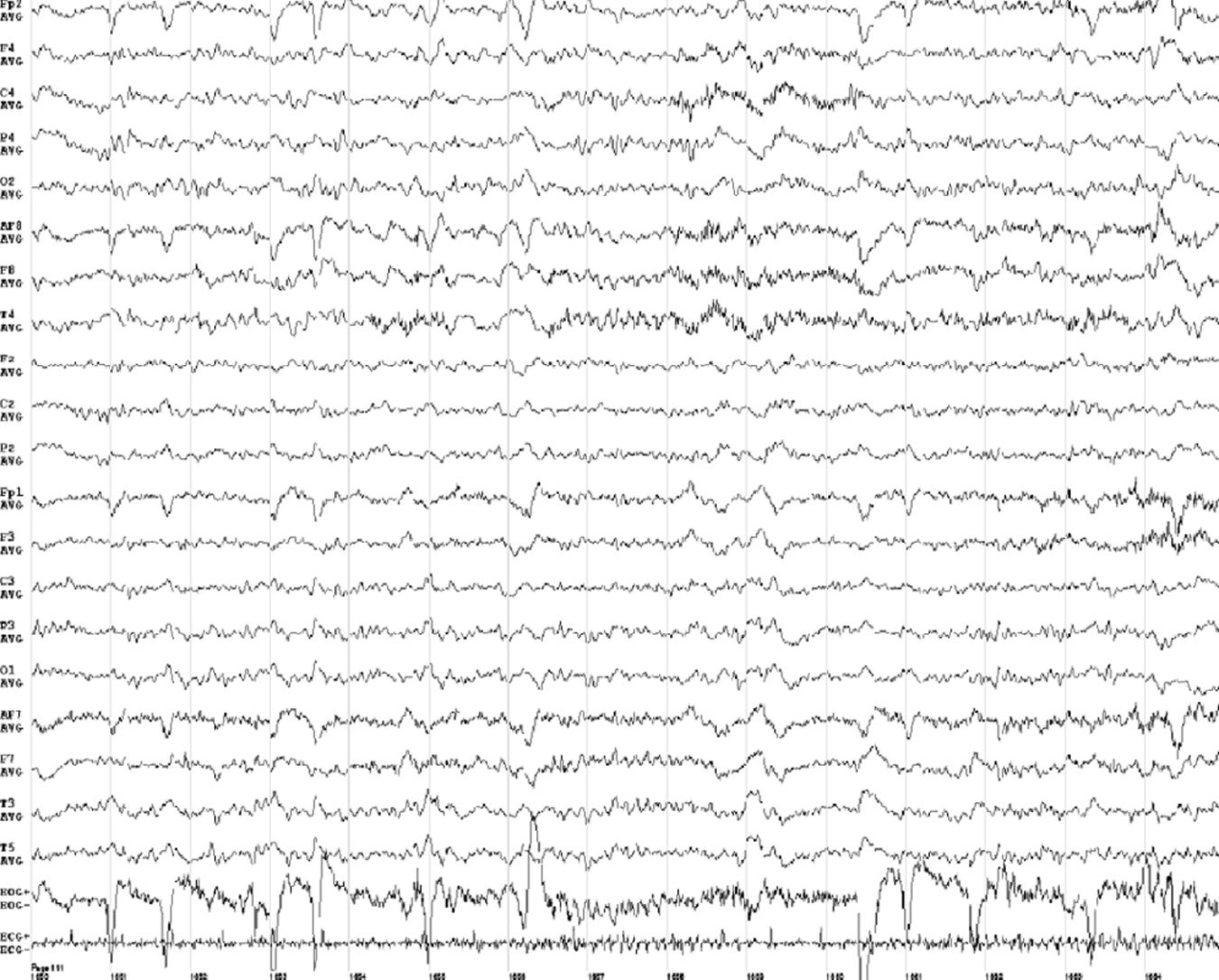
At one month of progression, we performed an awake EEG (Fig. 2), which revealed slow background activity, suggesting diffuse brain dysfunction (grades 2-3). Vitamin B1 level was 193ng/mL. The patient was transferred to another institution for recovery and continued with feeding via nasogastric tube.

From the second month of treatment, our patient presented good general appearance, fluent and coherent speech, an MMSE score of 23 (4 in orientation, 3 in registration, 4 in attention and calculation, 3 in recall, and 9 in language and copying); she was self-critical, interacted with her family, and could walk only with assistance. She needed help eating and with personal care. The patient participated in craft activities and regular rehabilitation sessions.
At 3 months, a significant improvement was observed in her nutritional status and she walked with a walker; a brain MRI scan revealed complete remission of the brain lesions (Fig. 1H-K).
The neuropsychological evaluation showed that autobiographical memory was preserved. We were able to apply only 3 subtests of the Wechsler Adult Intelligence Scale (WAIS-III): matrix reasoning, similarities, and digit span; results were higher level, average level, and average level, respectively. No areas of deficit were identified.
WE should be considered in the differential diagnosis of all patients with delirium8 or acute ataxia. Structural diseases of the medial thalamus, hippocampus, or the inferior medial region of the temporal lobe should also be considered due to the similar neuroanatomical involvement to WE. These include top of the basilar syndrome, hypoxic-ischaemic encephalopathy following cardiac arrest, herpes simplex encephalitis, and third ventricle tumour.9,10
Age of onset in our patient is atypical (eighth decade of life); she presented the classic triad of WE (encephalopathy, oculomotor dysfunction, and ataxia), although it was not observed at admission. In this case, WE was severe and was caused by malnutrition.11 She lost 33% of her body weight, with a BMI of 15.6 (BMI below 16 corresponds to grade 3/severe thinness according to the WHO classifications [1995,2000]).
Protein-calorie deficiency is not always present; in a review of 625 cases reported in the literature, the cause of WE was fasting or malnutrition in 10.2% of cases.12
Brain MRI showed the characteristic findings of WE, but this test is more sensitive for detecting WE lesions in non-alcoholic than in alcoholic patients13; clinical progression was excellent with vitamin supplementation.
Regarding the pathophysiology of these symptoms, thiamine reserves were depleted in 2-3 weeks due to caloric restriction. In the event of thiamine depletion, the function of the thiamine-dependent enzyme systems deteriorates and blood thiamine levels decrease. This damage occurs 4 days after onset of thiamine deficiency and eventually progresses to programmed cell death. At 14 days, brain lesions develop.11 It is probable that some subjects with genetically reduced transkelotase activity require higher levels of thiamine and therefore present a higher risk of WE in situations of increased demand or lower absorption.13 The low level of magnesium (a thiamine cofactor) also helped in the development of the disease. Other associated factors were vitamin B12 deficiency (long-term use of pantoprazole suppresses gastric acid production, which may lead to vitamin B12 malabsorption14), and vitamin D and albumin deficiency.
Our case is interesting as despite the several weeks of progression with altered mental state, vision, and gait, the first diagnostic hypothesis was vertebrobasilar stroke. Furthermore, WE was diagnosed in a context of severe malnutrition in a non-alcoholic patient, despite symptoms and neuroimaging findings being more typical of an alcoholic patient.
We propose that WE should be considered in patients of advanced age with altered level of consciousness of unknown cause, even in non-alcoholic patients; infusion of thiamine should be started immediately when the disorder is suspected, even in the absence of typical symptoms.
With this case, we aim to raise awareness of the need to identify this preventable, treatable, and high-mortality disease.
Please cite this article as: Ros Forteza FJ, Cabrera H, Bousende M. Desnutrición en el anciano y encefalopatía de Wernicke. Neurología. 2019;34:543–546.
