



Describir las características principales y el manejo clínico de una cohorte de pacientes con miopatía necrosante inmunomediada (MNIM).
MétodosEstudio observacional, unicéntrico y retrospectivo en el que se incluyeron los casos de MNIM diagnosticados en la Unidad de Neuromuscular de un hospital terciario en Madrid (España) entre los años 2013 y 2021.
ResultadosDieciséis pacientes con MNIM se incluyeron en el estudio, con una mediana de edad de 71,5 años (rango, 36-80), de los cuales 9 (56,3%) eran mujeres y 13(81,3%) habían sido tratados previamente con estatinas. El tiempo transcurrido desde el inicio de los síntomas hasta el diagnóstico fue menor de 6meses en 11 casos (68,8%) y, en todos, la forma de inicio fue la aparición de debilidad proximal en miembros inferiores, seguido de mialgias como segundo síntoma más frecuente. En 13pacientes los anticuerpos anti-3-hidroxi-3-metilglutaril coenzima A reductasa (HMGCR) fueron positivos, que fueron los únicos autoanticuerpos asociados a miositis detectados en la muestra. El abordaje terapéutico inicial se basó en el uso de corticoides, aunque la combinación con otros inmunosupresores fue necesaria desde el inicio en 8(50%) casos.
ConclusionesEl diagnóstico de MNIM dentro de las miopatías inflamatorias ha aumentado en los últimos años. En nuestra muestra los únicos anticuerpos detectados fueron los anti-HMGCR, una pieza clave en el diagnóstico diferencial con otras miopatías inflamatorias. El tratamiento de la enfermedad debe ser individualizado y tener en cuenta la respuesta inicial a corticoides, ya que puede requerir la combinación de varios fármacos.
To describe the main features and the clinical management of a cohort of patients with immune-mediated necrotizing myopathy (IMNM).
MethodsWe conducted an observational, monocentric, retrospective study of IMNM patients diagnosed in the Neuromuscular Unit of a tertiary hospital in Madrid (Spain) between 2013 and 2021.
ResultsSixteen IMNM cases were diagnosed, with a median age of 71.5 years (range 36-80), 9of whom (56.3%) were female. Thirteen (81.3%) patients had previously been treated with statins. The time from symptoms onset to diagnosis was less than 6months in 11 patients (68.8%) and the most common clinical symptoms were proximal weakness and myalgia. The only myositis specific autoantibodies detected were anti-3-hydroxy-3-methyl-coenzyme A reductase in 13 patients. The treatment strategy was based on prednisone, although a combination with other immunosuppressive drugs was required in 8(50%) patients.
ConclusionsThere has been an increase in the diagnosis of immune-mediated necrotizing myopathies in the last few years. The anti-HMGCR antibodies were the only ones detected in this sample, showing their key role in the diagnosis. Early recognition of the disease facilitates to start treatment as soon as possible, which should be based on the initial response to corticosteroids and usually requires a combination of several drugs.
Las miopatías inflamatorias idiopáticas (MII) constituyen un grupo heterogéneo de enfermedades cuya característica definitoria es la presencia de inflamación muscular que se acompaña de manifestaciones extramusculares que pueden afectar a la piel, los pulmones, el corazón y las articulaciones, y que se presentan con una frecuencia variable en los distintos tipos de MII1. Entre las pruebas de laboratorio, cabe destacar la asociación con autoanticuerpos específicos, que aparecen, aproximadamente, en el 60-70% de los pacientes con MII y resultan de gran utilidad para guiar el diagnóstico y para identificar diferentes grupos con características propias2,3.
Otra prueba fundamental en el diagnóstico de las MII es la biopsia muscular, ya que las distintas características histológicas sumadas a los datos clínicos y a la presencia de anticuerpos han permitido desarrollar un sistema para clasificar las diferentes MII en 4subgrupos principales: dernatomiositis, miositis asociada a síndrome antisintetasa, miositis por cuerpos de inclusión y miopatía necrosante inmunomediada (MNIM)4.
Dentro de estos subgrupos, la MNIM fue la última en ser descrita y, respecto al resto, se caracteriza por la presencia de mayor debilidad muscular y niveles más altos de creatinacinasa (CK), con manifestaciones extramusculares poco frecuentes5. El primer hito en la descripción de las MNIM fue la de los anticuerpos frente a la partícula de reconocimiento de señal (anti-SRP)6-8. Tras la publicación de varios estudios, empezó a llamarse «miositis anti-SRP» a un grupo de MII que se asociaban a estos anticuerpos. Poco después, se produjo la definición de las principales características histológicas con fibras necróticas y escasos infiltrados inflamatorios, lo que llevó a considerar las MNIM una entidad separada entre las MII en 20039. En 2010, se detectó un nuevo autoanticuerpo, que tiene como diana la enzima 3-hidroxi-3-metilglutaril coenzima A reductasa (anti-HMGCR) en pacientes con diagnóstico de MNIM10. Esta enzima cataliza reacciones clave en la biosíntesis del colesterol y su expresión en la membrana celular está aumentada en pacientes tratados con estatinas, lo que puede suponer un mecanismo importante en la fisiopatología de la enfermedad, aunque los mismos anticuerpos se han detectado también en pacientes que nunca han sido tratados con fármacos hipolipidemiantes11. Por último, existe un tercer subgrupo dentro de los pacientes con MNIM, que representa en torno al 40% de los casos, en los que no se detecta ningún autoanticuerpo12.
El abordaje terapéutico de los pacientes con MNIM se basa en series de casos y consensos de expertos e incluye, además de los corticoides, varios fármacos inmunosupresores e inmunomudulares, cuyo uso en combinación suele requerirse para controlar la enfermedad13. Además, será necesario el despistaje de un posible cáncer subyacente, que se ha relacionado con formas seronegativas, y, de forma más controvertida, con MNIM asociada a anti-HMGCR14.
MétodosTras una revisión retrospectiva de la base de datos de la Unidad de Neuromuscular del Hospital General Universitario Gregorio Marañón (Madrid), se identificaron los casos de MNIM diagnosticados entre los años 2013 y 2021. Los criterios de inclusión se adaptaron del último consenso de clasificación clinicoseropatológica del European Neuromuscular Centre (ENMC)13. Además de una biopsia muscular compatible con fibras necróticas o regenerativas en ausencia de infiltrado inflamatorio o con un infiltrado inflamatorio escaso se requirieron niveles de CK elevados en presencia de un cuadro clínico de debilidad muscular de reciente aparición13.
Los criterios de exclusión fueron no cumplir todos los criterios de inclusión, historia familiar de miopatía hereditaria con causa genética conocida y que la biopsia muscular o el resto de las pruebas complementarias fuesen sugestivas de otra causa alternativa.
En cada caso se revisó la historia clínica completa del paciente y se recogieron los datos demográficos, los datos clínicos y la fecha del diagnóstico. Con base en el tiempo transcurrido desde el inicio de síntomas, se clasificaron en: inicio agudo (menos de 4semanas), inicio subagudo (entre 4semanas y 6meses) o crónico (si habían transcurrido más de 6meses desde el inicio). Se valoró el patrón de la debilidad muscular (miembros inferiores vs. miembros superiores, proximal vs. distal, simétrico vs. asimétrico) y el grado de gravedad se midió mediante la escala del Medical Research Council (MRC) y fue considerada grave si había una puntuación MRC≤3/5 en alguno de los grupos musculares explorados.
A continuación se evaluaron las pruebas complementarias realizadas tras la primera valoración en la consulta de neurología y se recogieron los valores de CK y los resultados del estudio de autoinmunidad. Los anticuerpos específicos de miositis tipo inmunoglobulina G (IgG) se testaron en suero utilizando una técnica de dot-blot (MYO12D-24, D-tek, Mons, Bélgica), que permite la detección y la medición semicuantitativa de 12 autoanticuerpos: antihistidil-ARN-t-sintetasa (Jo1), antitreonina-ARN-t-sintetasa (PL7), antialanina-ARN-t-sintetasa (PL-12), antiglicil-ARN-t-sintetasa (EJ), anti-SRP, antihistona-desacetilasa remodeladora de nucleosomas (Mi2), antigén 5 asociado a la diferenciación de melanoma (MDA5), antifactor intermediario de transcripción 1-γ (TIF1-γ), anti-HMGCR, antiantígeno A relacionado con el síndrome de Sjogren, autoanticuerpos (SSA/Ro52kD), antienzima de activación del modificador similar a la ubiquitina pequeña 1/2 (SAE 1/2) y antiproteína 2 de la matriz nuclear (NXP2).
En todos los casos se realizó un electromiograma. El estudio fue llevado a cabo por el Servicio de Neurosifiología e incluyó una evaluación tanto de la actividad de reposo para valorar la presencia de actividad espontánea como de la actividad durante la contracción leve y máxima, para el análisis de los potenciales de unidad motora (PUM). El número de músculos explorados en cada paciente se basó en el criterio clínico del médico que realizaba la prueba, incluyendo músculos proximales y distales en miembros superiores (trapecio, deltoides, flexor radial del carpo, bíceps braquial, primer interóseo dorsal, flexor largo de los dedos) y en miembros inferiores (ileopsoas, glúteo medio, tensor de la fascia lata, recto femoral, vasto lateral, bíceps femoral, tibial anterior y gastrocnemio lateral). No se realizaron estudios neurofisiológicos de control.
En los casos en los que estaba disponible, se revisaron las imágenes de resonancia magnética (RM) muscular de cuerpo entero, incluyendo secuencias spin eco turbo (TSE) potenciadas en T1 y secuencias de recuperación de inversión tau corta potenciadas en T2 (STIR).
Las biopsias musculares, obtenidas en el momento del diagnóstico, fueron procesadas y evaluadas por el Servicio de Anatomía Patológica. En primer lugar, las muestras fueron orientadas y criopreservadas a −80°C, mientras que un pequeño fragmento se preservó en glutaraldehído para el análisis ultraestructural. Las tinciones rutinarias incluyeron: hematoxilina-eosina (HE), tricrómico de Gomori, PAS, OIL-RED O, ATPase 42, ATPase 46, ATPase 94, DPNH 30, succinato y succinato-COX. Posteriormente se llevaron a cabo procedimientos de inmunohistoquímica para completar el estudio de miopatía inflamatoria, que incluyeron el marcaje de CD3, CD20, CD68, p62 y HLA-I. Los anticuerpos utilizados, de acuerdo con los protocolos de tinción del fabricante, fueron: anticuerpos anti-CD3 (Agilent, polyclonal rabbit anti-human), anti-CD20 (Agilent, monoclonal mouse anti-human clone L26), anti-CD68 (Agilent, monoclonal mouse anti-human clone PG-M1), anti-p62 (BD Transduction Laboratories, purified mouse anti-human clone 45/p62[dok]) y anti-HLA-I (Agilent, monoclonal mouse anti-human clone W6/32).
En cuanto al manejo clínico de los pacientes con MNIM, se recogieron todos los datos sobre la administración de distintos fármacos inmunosupresores, con los detalles de las diferentes pautas y si se habían empleado o no de forma combinada. Las estrategias terapéuticas se clasificaron en función del número de tratamientos administrados en: monoterapia (un único fármaco), biterapia (2 fármacos) o politerapia (3 o más fármacos).
La respuesta clínica al tratamiento fue reevaluada 6meses después en el seguimiento, tanto de forma subjetiva (mejoría, empeoramiento o estabilización de síntomas) como de forma objetiva mediante el balance muscular medido por la escala MRC. A los 6meses se recogieron nuevamente los niveles de CK y se midió el grado de discapacidad utilizando la escala modificada de Rankin15. Se reportaron todas las recaídas durante el tiempo de seguimiento del estudio y el diagnóstico de cáncer asociado a miositis se consideró en pacientes en los que el cáncer había sido diagnosticado en los 3años siguientes al diagnóstico de MNIM, siguiendo los criterios previos utilizados para el diagnóstico de cáncer asociado a miositis16.
Este estudió recibió la aprobación del Comité de Ética para la Investigación con Medicamentos del Instituto de Investigación Sanitaria del Hospital General Universitario Gregorio Marañón.
Análisis estadísticoEl análisis estadístico se llevó a cabo utilizando el programa SPSS® (Statistical Package for the Social Sciences), versión 21. Las variables categóricas se definieron como número de casos y porcentajes y las variables continuas mediante rangos y medianas.
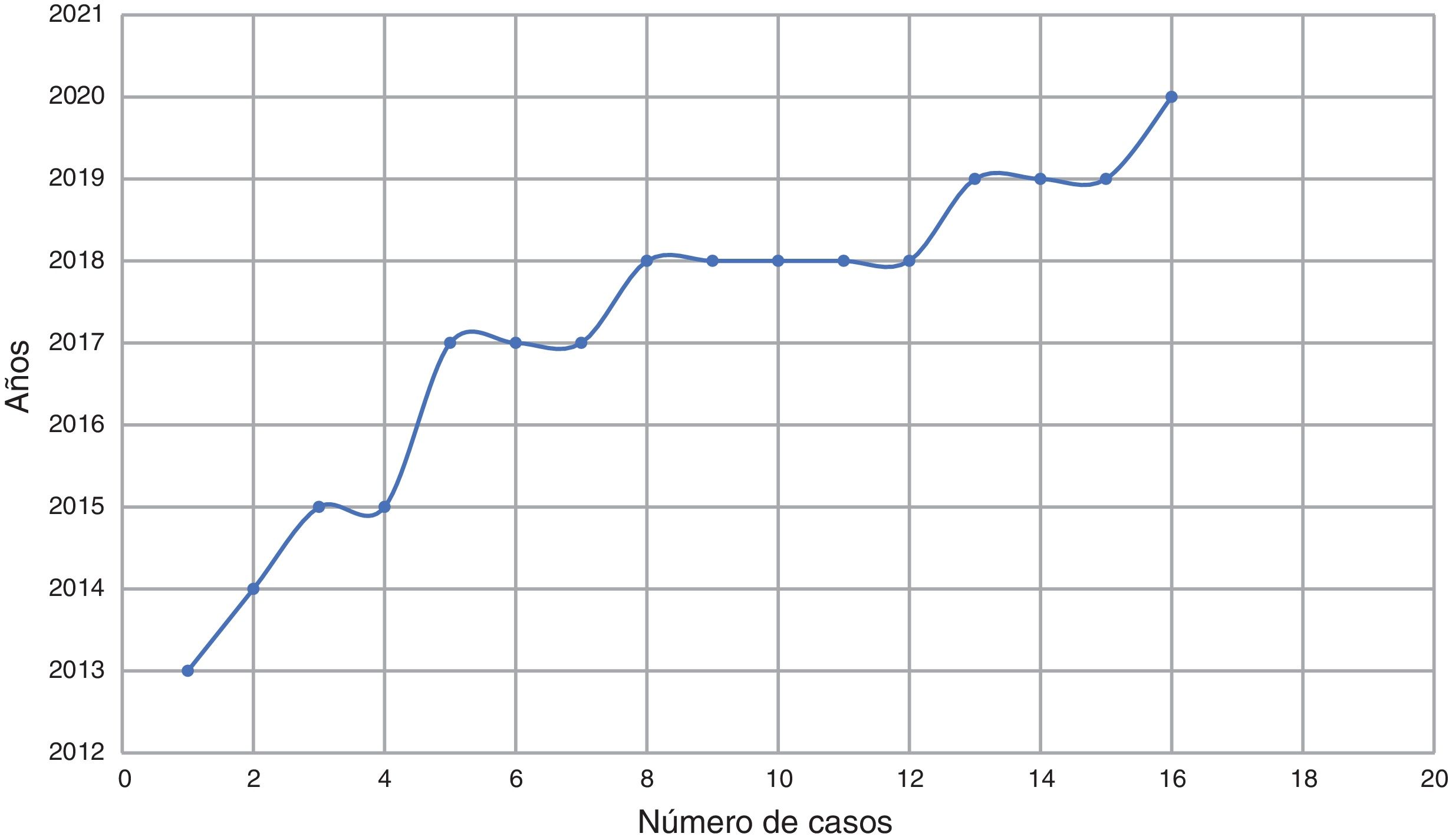
ResultadosDieciséis pacientes cumplieron todos los criterios y se incluyeron en el estudio. Lo primero destacable fue la tendencia creciente en el diagnóstico de MNIM en nuestra muestra, con un número de pacientes diagnosticados por año que fue aumentando progresivamente del 2013 al 2021 (fig. 1).

La mediana de edad para el inicio de los síntomas fueron 71,5 años (rango 36-80) y 9pacientes (56,3%) eran mujeres. Antes del inicio de los síntomas, 13pacientes (81,3%) habían recibido tratamiento con estatinas, de las cuales la atorvastatina fue la más frecuente (utilizada en 9pacientes; 62,5%) seguida de simvastatina y rosuvastatina, en 2pacientes (12,5%) cada una.
El tiempo transcurrido desde el inicio de síntomas hasta el diagnóstico fue inferior a 6meses en 11pacientes (68,8%); la forma de presentación subaguda, en 9 casos (56,3%), fue la más frecuente.
La debilidad muscular proximal de extremidades fue el síntoma inicial en todos los pacientes y fue considerada grave en 10(62,5%). La distribución fue simétrica en 15pacientes (93,8%) y más pronunciada en miembros inferiores en 11pacientes (68,8%) mientras que en 5(31,2%) afectó de forma similar a miembros superiores e inferiores. El segundo síntoma más frecuente fueron las mialgias (12pacientes; 75%), seguidas de debilidad cervical y disfagia, presentes en 4pacientes (25%) cada una.
En 2casos (11,8%) la debilidad muscular se asoció a otras manifestaciones extramusculares relacionadas con el diagnóstico previo de otras enfermedades inmunomediadas: una nefropatía membranosa con un síndrome antifosfolípido secundario a una enfermedad autoinmune no filiada en un caso y una vasculitis sistémica en el otro.
Pruebas de laboratorio complementariasEl valor de la CK alcanzó una mediana de 4.746 unidades internacionales por litro (UI/L) con un rango entre 1.001 y 9.856 UI/L.
Tras los estudios de autoinmunidad para investigar la presencia de autoanticuerpos específicos de miositis, se detectaron los anticuerpos anti-HMGCR en 13pacientes (81,3%), de los cuales 11(84,6%) estaban recibiendo tratamiento con estatinas antes de los síntomas, mientras que, en el grupo de los 3pacientes seronegativos, 2(66,7%) tomaban estatinas. En ningún paciente se encontraron los anticuerpos anti-SRP también asociados a MNIM. Los pacientes seronegativos incluyeron los 2casos con manifestaciones extramusculares asociadas a enfermedad autoinmune sistémica y el de una paciente que se presentó con el cuadro clínico típico de la enfermedad.
En cuanto al resto de los estudios analíticos, cabe destacar que los anticuerpos antinucleares fueron positivos en 2casos (12,5%), asociados a vasculitis sistémica en una paciente y de forma concomitante a anti-HMGCR en el otro.
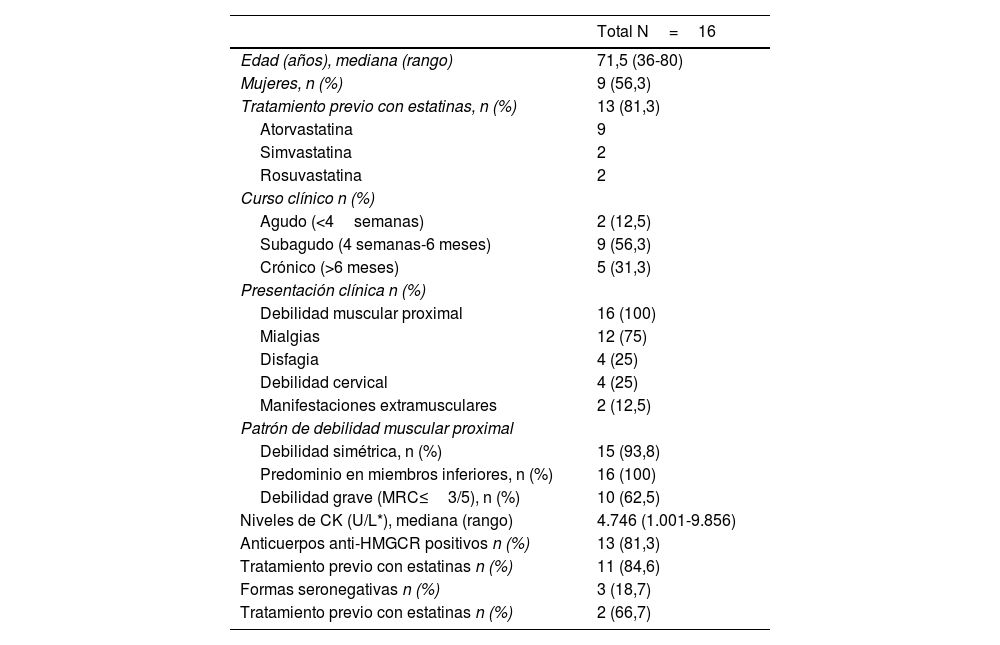
Los resultados de las principales pruebas de laboratorio se resumen en la tabla 1 junto a las principales características demográficas y clínicas.
Principales características demográficas, clínicas y pruebas de laboratorio p en 16pacientes con MNIM
| Total N=16 | |
|---|---|
| Edad (años), mediana (rango) | 71,5 (36-80) |
| Mujeres, n (%) | 9 (56,3) |
| Tratamiento previo con estatinas, n (%) | 13 (81,3) |
| Atorvastatina | 9 |
| Simvastatina | 2 |
| Rosuvastatina | 2 |
| Curso clínico n (%) | |
| Agudo (<4semanas) | 2 (12,5) |
| Subagudo (4 semanas-6 meses) | 9 (56,3) |
| Crónico (>6 meses) | 5 (31,3) |
| Presentación clínica n (%) | |
| Debilidad muscular proximal | 16 (100) |
| Mialgias | 12 (75) |
| Disfagia | 4 (25) |
| Debilidad cervical | 4 (25) |
| Manifestaciones extramusculares | 2 (12,5) |
| Patrón de debilidad muscular proximal | |
| Debilidad simétrica, n (%) | 15 (93,8) |
| Predominio en miembros inferiores, n (%) | 16 (100) |
| Debilidad grave (MRC≤3/5), n (%) | 10 (62,5) |
| Niveles de CK (U/L*), mediana (rango) | 4.746 (1.001-9.856) |
| Anticuerpos anti-HMGCR positivos n (%) | 13 (81,3) |
| Tratamiento previo con estatinas n (%) | 11 (84,6) |
| Formas seronegativas n (%) | 3 (18,7) |
| Tratamiento previo con estatinas n (%) | 2 (66,7) |
El electromiograma demostró PUM miopáticos (duración corta, con baja amplitud y polifasia) en músculos proximales en 15pacientes (93,8%). Este patrón miopático se acompañó de la presencia de actividad espontánea en reposo en 12casos (75%) y se registró en forma de fibrilaciones, ondas positivas y descargas repetitivas complejas.
En 3pacientes (18,8%) se detectaron descargas miotónicas en ausencia de miotonía clínica, en 2de ellos solo en el vasto lateral, mientras que el otro se detectó de forma difusa en el bíceps braquialis, glúteo medio, recto femoral e ileopsoas.
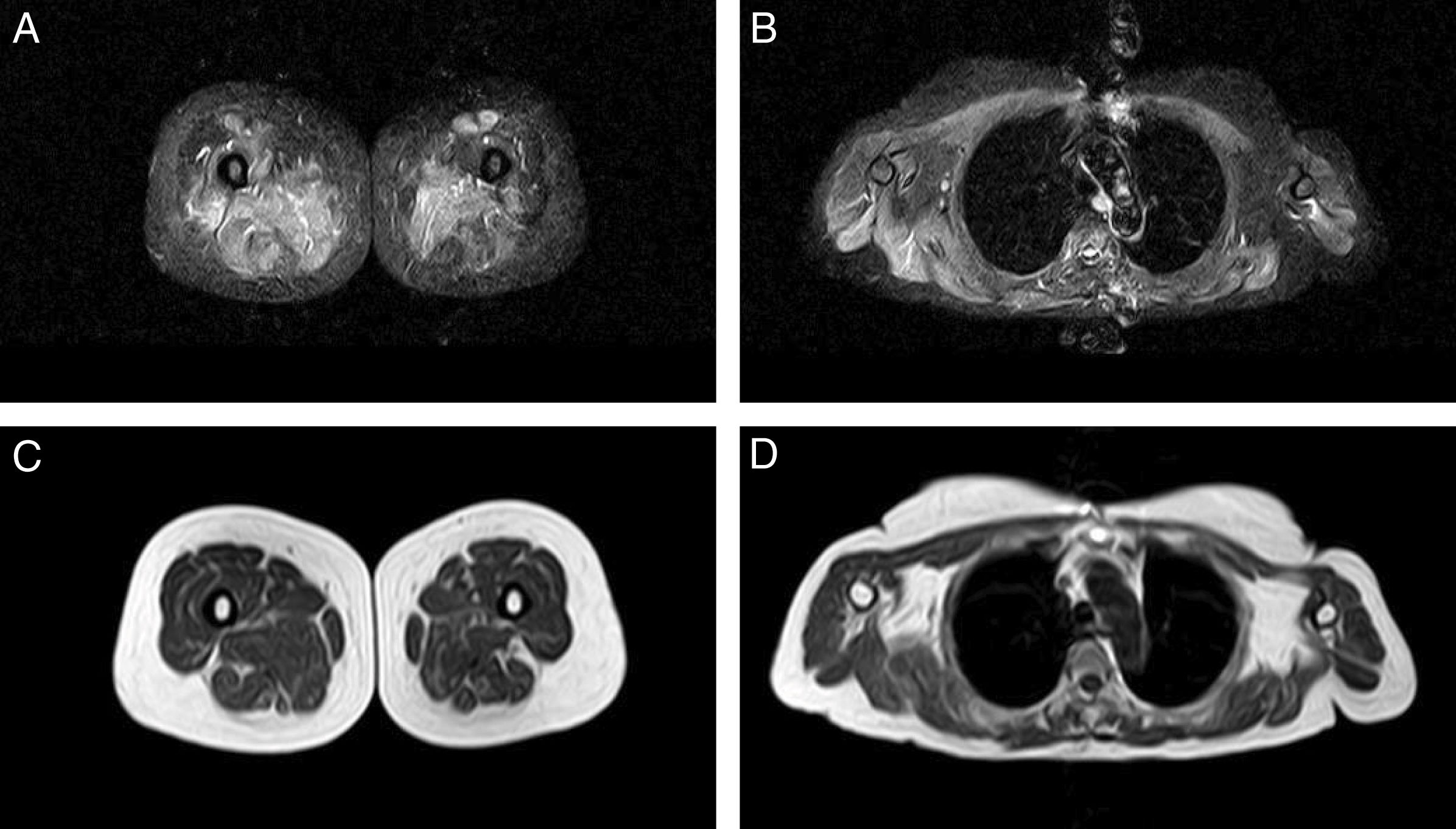
Resonancia magnética muscularLa RM muscular de cuerpo entero se realizó únicamente en un paciente, ante dudas diagnósticas, debido a una forma de presentación atípica con síntomas leves. En las secuencias STIR, se demostró la presencia de aumento de señal compatible con edema muscular, sin observarse sustitución grasa en secuencias T1 (fig. 2). Los músculos más afectados fueron los glúteos en la cintura pélvica, los músculos aductores en el compartimento medial del muslo, el bíceps femoral en el compartimento posterior y los vastos lateral e intermedio, así como el recto femoral, en el compartimento anterior. De forma menos llamativa se objetivó también edema muscular en los músculos periescapulares y en el deltoides.
 (A), que afecta a los músculos aductores en compartimento medial del muslo, vasto lateral y recto femoral en el compartimento anterior y bíceps femoral en el compartimento posterior, más prominentes en el lado derecho. B) En la cintura escapular se objetivó un aumento de señal en músculos periescapulares, deltoides y, de forma menos llamativa, en músculos pectorales, de predominio en el lado derecho. Estos cambios no se acompañaron de sustitución grasa en secuencias spin eco turbo (TSE) potenciadas en T1 (C y D).")
En una RM muscular de cuerpo entero, en los muslos se apreció aumento de señal en secuencias de recuperación de inversión tau corta potenciadas en T2 (STIR) (A), que afecta a los músculos aductores en compartimento medial del muslo, vasto lateral y recto femoral en el compartimento anterior y bíceps femoral en el compartimento posterior, más prominentes en el lado derecho. B) En la cintura escapular se objetivó un aumento de señal en músculos periescapulares, deltoides y, de forma menos llamativa, en músculos pectorales, de predominio en el lado derecho. Estos cambios no se acompañaron de sustitución grasa en secuencias spin eco turbo (TSE) potenciadas en T1 (C y D).
Los 16 pacientes (100%) de la muestra presentaron las fibras necróticas y regenerativas definitorias de la enfermedad en los estudios de anatomía patológica.
Se acompañaron de un infiltrado inflamatorio en 8casos (50%), 6pacientes (37,5%) presentaban infiltrados inflamatorios linfocitarios poco prominentes, mientras que en 3casos (18,7%) las fibras necróticas se asociaron a infiltrados macrofágicos. El inmunomarcaje del HLA de tipo 1 se llevó a cabo en 14pacientes y fue positivo en 9pacientes (64,3%), en los que se distribuyó en el sarcolema y el citoplasma de las fibras no necróticas.
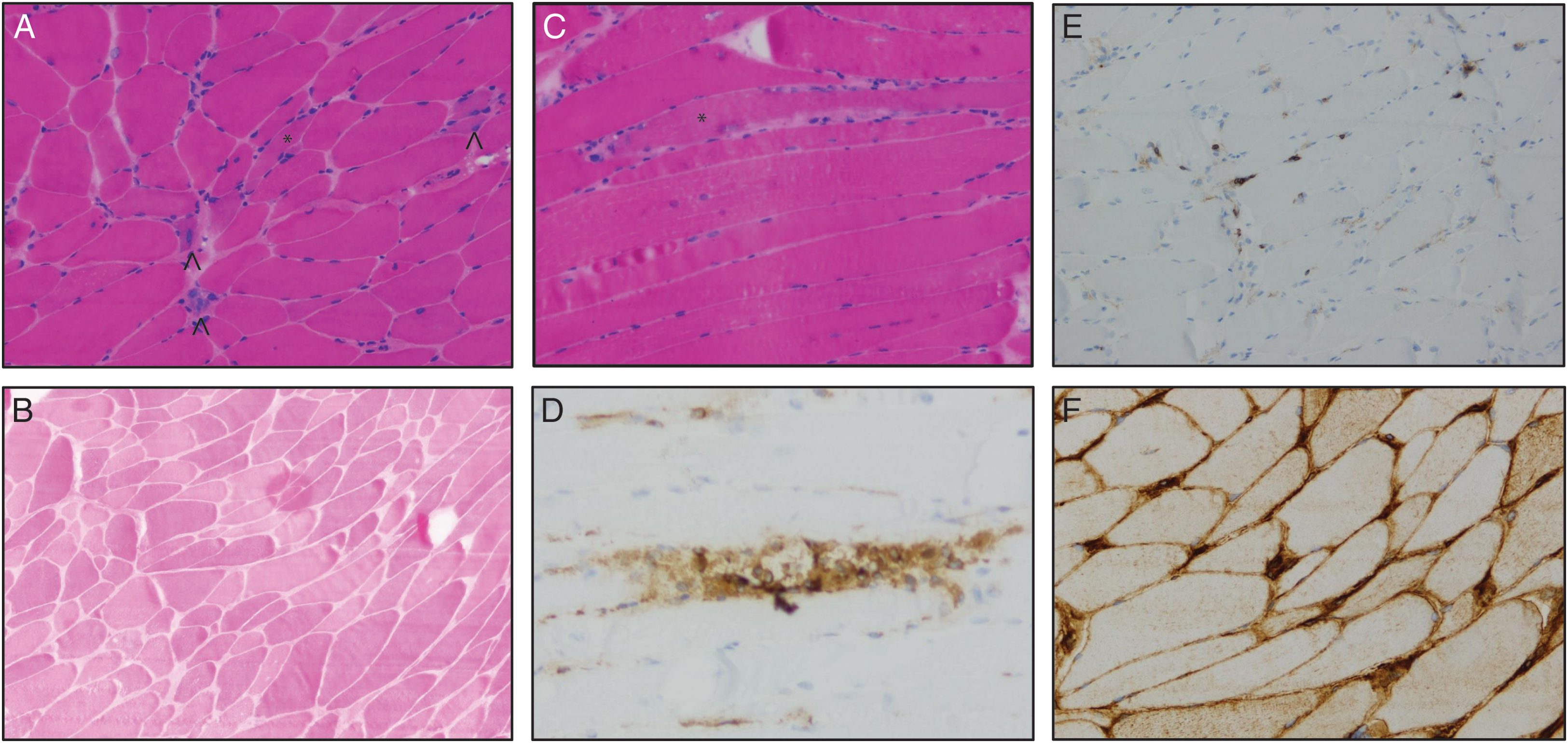
En la figura 3 se muestra un ejemplo de las principales características histológicas de los pacientes de nuestra muestra.
 con grupos de fibras anguladas atróficas (asterisco), fibras regenerativas (puntas de flecha) y ocasional miofagocitosis (b; asterisco) en tinciones de hematoxilina-eosina (H-E). La tinción mediante ATPasa 4,2 demostró la presencia de fibras de diferentes tipos (b). En tinciones de inmunomarcaje se observan macrófagos CD-68 positivos en endomisio y en el interior de las fibras musculares (d), linfocitos CD-3 positivos dispersos (e) y tinción positiva para HLA de tipo 1 con tinción intensa de la membrana y pequeña extensión al sarcoplasma (f).")
Biopsia muscular. Las imágenes muestran varios cortes de músculo esquelético estriado en las que se objetiva moderada variabilidad en el tamaño de fibras (a) con grupos de fibras anguladas atróficas (asterisco), fibras regenerativas (puntas de flecha) y ocasional miofagocitosis (b; asterisco) en tinciones de hematoxilina-eosina (H-E). La tinción mediante ATPasa 4,2 demostró la presencia de fibras de diferentes tipos (b). En tinciones de inmunomarcaje se observan macrófagos CD-68 positivos en endomisio y en el interior de las fibras musculares (d), linfocitos CD-3 positivos dispersos (e) y tinción positiva para HLA de tipo 1 con tinción intensa de la membrana y pequeña extensión al sarcoplasma (f).
Tras el diagnóstico de MNIM, se inicio tratamiento farmacológico en todos los pacientes. El abordaje inicial se basó en el uso de prednisona a dosis de 1mg/kg. En esta primera fase, de tratamiento de inducción, la prednisona a dosis de 1mg/kg fue el primer fármaco utilizado en todos los casos, menos en una paciente con múltiples comorbilidades, incluidas diabetes mellitus de mal control y mala situación funcional previa, en la que se inició tratamiento con azatioprina.
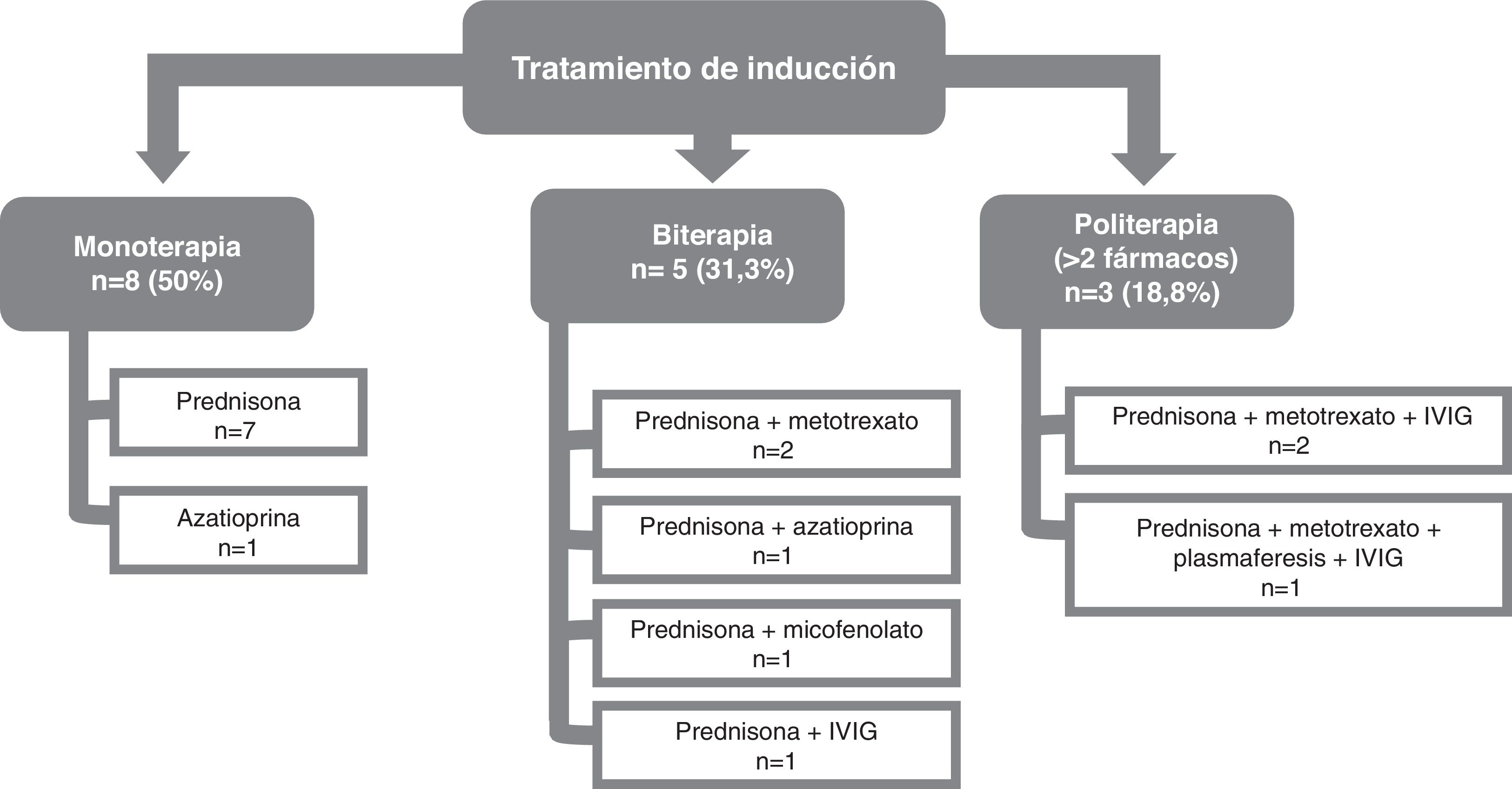
Con base en la respuesta inicial a prednisona y la gravedad de la afectación clínica, se requirió el uso de otros fármacos durante las primeras 6semanas tras el diagnóstico, 6pacientes precisaron el uso de biterapia (31,3%) y 3pacientes (18,8%) politerapia con 3fármacos. Los tratamientos empleados en combinación con mayor frecuencia fueron las inmunoglobulinas intravenosas (IGIV) en 5pacientes y otros fármacos inmunosupresores, como metotrexato (en 5pacientes), azatioprina (en 2pacientes) y micofenolato de mofetilo (en un paciente). En un caso se procedió al recambio plasmático mediante 8sesiones de plasmaféresis en días alternos, ante la clínica grave y la ausencia de mejoría tras el inicio de prednisona de 1mg/kg y tras asociar metotrexato. El paciente, un varón de 71 años con anticuerpos anti-HMGCR positivos y una biopsia muscular en la que presentaba un pequeño infiltrado inflamatorio linfocitario y macrofágico asociado a las fibras necróticas en ausencia de HLA-1, no presentó tampoco ninguna mejoría durante el recambio plasmático ni en los 10días siguientes, por lo que finalmente se le añadió IGIV al tratamiento, con una lenta mejoría en las semanas siguientes.
Los distintos esquemas terapéuticos empleados como tratamiento de inducción se detallan en la figura 4.

Tras el tratamiento de inducción, 6meses después del diagnóstico, 13pacientes referían mejoría clínica subjetiva (81,3%) y la mediana de CK disminuyó a 420 UI/L (rango, 27-1.719 UI/L); 7 pacientes (43,8%) alcanzaron una resolución completa de la debilidad, 4(25%) experimentaron una mejoría clínica significativa de la fuerza muscular y en 2 casos (12,5%) los síntomas permanecieron sin cambios. En 3pacientes (18,7%) el grado de debilidad continuaba siendo grave.
Entre los posibles factores relacionados con la respuesta clínica inicial al tratamiento, se consideró como una buena respuesta clínica alcanzar una resolución completa o una mejoría clínica significativa. Los estudios neurofisiológicos mostraron una proporción algo mayor de pacientes (9 casos; 75%) con resolución de síntomas o mejoría significativa, en el grupo de pacientes con actividad espontánea frente a los que no la presentaban (2casos; 50%). En cuanto a la anatomía patológica, una buena respuesta clínica se alcanzó con mayor frecuencia en el grupo con HLA de tipo positivo (8pacientes; 88,9%) frente al negativo (3; 60%); mientras que para la presencia o no de infiltrado inflamatorio fue similar, con 5casos (62,5%) en el grupo con infiltrado inflamatorio y 6casos (75%) en el grupo que no asociaba infiltrado inflamatorio.
De los 3pacientes seronegativos, que fueron los que asociaron manifestaciones extramusculares, ningunó presentó un grado de debilidad grave al diagnóstico, y tuvieron una buena respuesta inicial al tratamiento de monoterapia, mientras que en el caso restante, aunque el grado de debilidad no fue grave al diagnóstico, sí requirió asociar metotrexato durante el tratamiento de inducción, con lo que se consiguió una buena respuesta clínica a los 6meses.
El tratamiento de mantenimiento fue diferente en función de la evolución clínica. En los casos en los que no hubo mejoría o no se alcanzó un grado de mejoría suficiente, fue necesario un esquema más agresivo de tratamiento, por lo que en ese momento 4pacientes (25%) se encontraban recibiendo politerapia con 3fármacos. En el caso de una mujer de 36 años, con anticuerpos anti-HMGCR sin previo tratamiento con estatinas que presentaba un curso lento, pero sin mejoría a pesar de tratamiento con prednisona y metotrexato, se inicio tratamiento con rituximab.
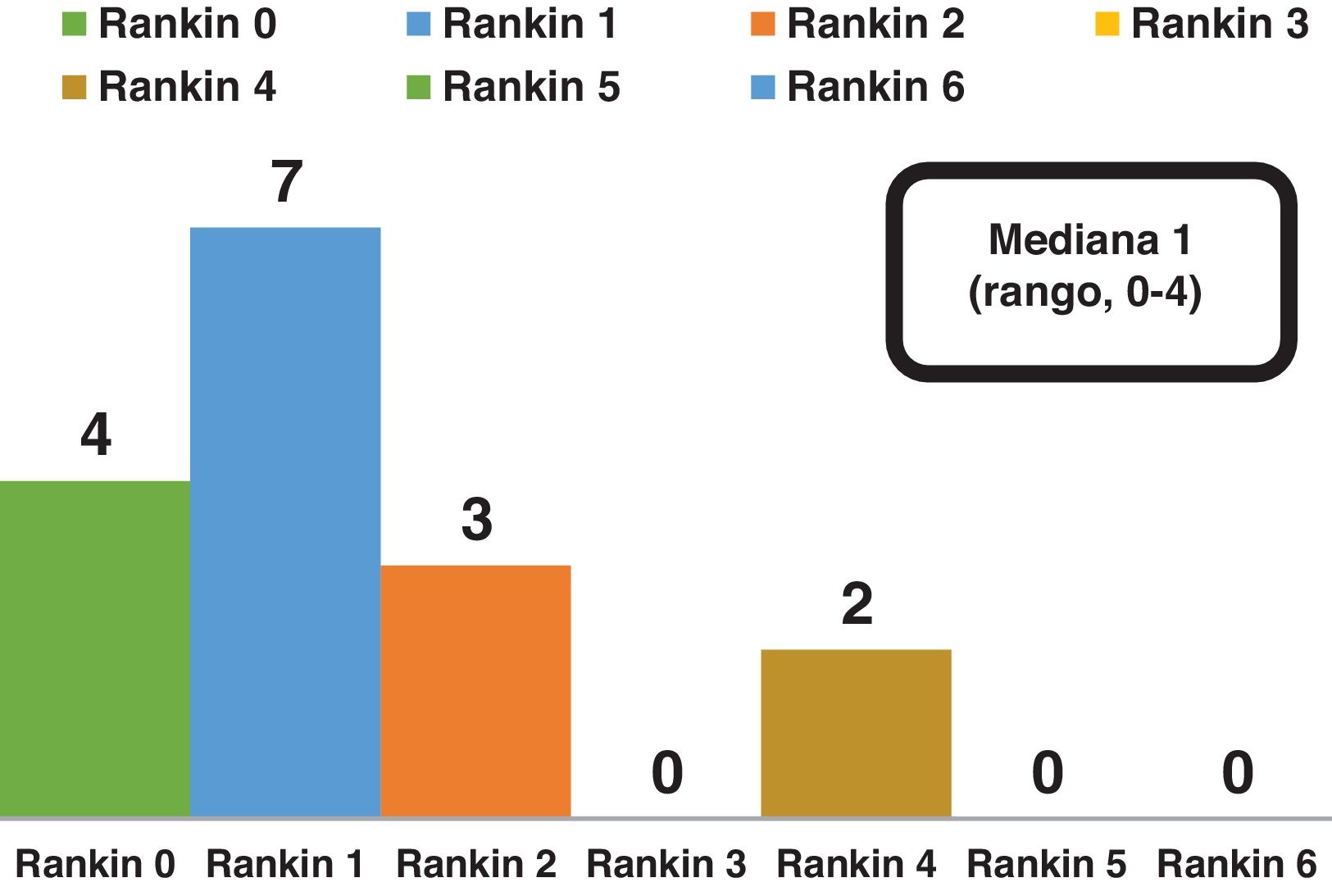
El grado de discapacidad medido mediante la escala modificada de Rankin a los 6meses alcanzó una mediana de 1 con un rango entre 0 y 4 (fig. 5).

Durante todo el tiempo de seguimiento del estudio, 2pacientes (12,5%) tuvieron una recaída y, en el caso de la paciente tratada con rituximab, los síntomas no mejoraron: tuvo una evolución muy lenta a pesar de haber utilizado varias líneas de tratamiento.
Por último, se detectó un caso de cáncer asociado a MNIM en un varón de 73 años con anticuerpos anti-HMGCR positivos al que se le diagnosticaron un liposarcoma y un cáncer escamoso lingual de forma concomitante a la miositis. El paciente fue intervenido quirúrgicamente de ambos tumores y recibió tratamiento con 3fármacos (prednisona, metotrexato e IGIV), a pesar de lo cual no hubo mejoría de síntomas musculares y falleció 9meses tras el inicio de los síntomas.
DiscusiónEste estudio retrospectivo describe las principales características de una cohorte de pacientes con diagnóstico de MNIM, incluyendo datos clínicos, autoanticuerpos, el patrón histológico, la estrategia de tratamiento y la evolución centrada, sobre todo, en los primeros 6meses tras el diagnóstico.
El primer dato importante obtenido de la muestra es que, a lo largo de los años del estudio, el diagnóstico de MNIM es cada vez más frecuente (fig. 1), lo que convierte a este subtipo de MII en una enfermedad emergente.
En línea con reportes previos5,12,13,17-19, la prevalencia de la enfermedad fue ligeramente superior en mujeres (56,3%), aunque la edad de inicio en nuestra muestra (mediana: 71,5 años) fue algo superior a otras series. Una posible razón es la alta prevalencia en esta cohorte de casos con anti-HMGCR positivos y la ausencia de casos con anticuerpos anti-SRP, habitualmente más jóvenes, y la alta prevalencia de casos con exposición previa a estatinas, habitualmente empleadas en adultos de mayor edad5,20.
En relación con los datos clínicos, la forma de presentación más frecuente fue aguda-subaguda, con debilidad muscular de extremidades proximal y simétrica, y niveles muy elevados de CK5,18. Sin embargo, merece la pena señalar un caso con anticuerpos anti-HMGCR positivos que cursó de forma lenta imitando a una distrofia muscular de cinturas, de forma similar a otros casos reportados, y que pone de manifiesto la importancia de solicitar una determinación de anticuerpos anti-HMGCR en casos con un diagnóstico de probable distrofia muscular de cinturas en los que no se ha determinado una causa genética o no existe historia familiar21,22. Otro dato clínico que tener en cuenta es que, de forma similar a estudios previos, las manifestaciones extramusculares fueron poco frecuentes, incluyendo una vasculitis sistémica y un síndrome antifosfolípido secundario a otra enfermedad autoinmune, lo que apoya la asociación entre algunas formas de MNIM y otras enfermedades sistémicas inmunomediadas que podrían suponer un factor de riesgo para el desarrollo de MNIM23. La evolución de estos pacientes fue favorable.
Continuando con los autoanticuerpos asociados a miositis, es relevante la ausencia de casos con anticuerpos anti-SRP y la alta prevalencia de formas asociadas a anticuerpos anti-HMGCR (81,3%). Una posible explicación podría ser el elevado número de pacientes que reciben tratamiento con estatinas en nuestro medio11,24-27. No obstante, el antecedente de tratamiento previo con estatinas en pacientes con MNIM asociada a anticuerpos anti-HMGCR varía en las diferentes series y en nuestra muestra el antecedente de tratamiento con estatinas también se reportó en 2pacientes (66,7%) de los casos seronegativos, lo que apoya que otros factores inmunogenéticos y medioambientales estén implicados en la fisitopatología de le enfermedad28-30.
Los estudios neurofisiológicos mostraron el patrón habitual con PUM miopáticos y actividad espontánea31 y el número de pacientes que asoció miotonía eléctrica (3; 18,8%) fue menor que en otras series. Una posible explicación podría ser que no se examinaron los músculos paraespinales, en los que con frecuencia se detecta la miotonía eléctrica en pacientes con MNIM, y que llegan a ser los únicos afectos en algunos casos. Los 3pacientes tenían anticuerpos anti-HMGCR y antecedente de exposición a estatinas, en consonancia con otros estudios que indican que, aunque la causa de la miotonía es desconocida, podría verse favorecida por el tratamiento previo con estatinas32.
Solo en un caso se completó el estudio mediante una RM muscular, debido a un cuadro clínico poco florido, que demostró la presencia de edema en ausencia de sustitución grasa. A diferencia de otros casos reportados en la literatura en los que es frecuente la presencia de infiltración grasa, en este caso puede no haberse detectado, al tratarse de un cuadro leve dentro del espectro de gravedad de las MNIM y haberse realizado la RM muscular de forma precoz en el curso de la enfermedad33.
Aunque se ha indicado la posibilidad de llegar al diagnóstico de MNIM sin una biopsia muscular en pacientes anti-HMGCR positivos34,35, dado que es necesaria en pacientes seronegativos y con el objetivo de no retrasar el inicio del tratamiento en espera de los resultados de los anticuerpos, se tomó dicha biopsia de forma rutinaria en todos los pacientes ante la sospecha clínica de MNIM.
El tratamiento, dada la ausencia de ensayos clínicos controlados y de guías clínicas, se basó en la experiencia clínica y en series retrospectivas de casos según las recomendaciones del último consenso de la ENMC13. Siguiendo estas pautas, el tratamiento inicial fueron los corticoides en todos los pacientes, a excepción de un caso en el que no se administraron por comorbilidad. Con base en la respuesta inicial, si no se conseguía una mejoría clínica se añadía un segundo o tercer fármaco (IGIV o metotrexato)5,12,18,20,36, que fueron las opciones que se emplearon con mayor frecuencia en nuestra muestra, pautados en 5pacientes cada uno y en 3casos utilizados en combinación. Las IGIV se recomiendan como parte de la terapia de inducción en enfermedad refractaria, incluso en monoterapia si existe contraindicación para el uso de corticoides. En esta serie no fueron utilizadas en ningún caso en monoterapia, pero sí se emplearon como segundo fármaco si no había una mejoría suficiente con corticoides y fueron consideradas en primer lugar tras corticoides si no había ninguna mejoría en pacientes con debilidad grave, en busca de una respuesta más precoz que con otros inmunosupresores.
Otros fármacos ahorradores de corticoides, como azatioprina, micofenolato y ciclosporina, se han propuesto como posibles alternativas, mientras que el rituximab se contempla como una opción terapéutica para pacientes refractarios con mala respuesta a tratamientos previos5,12,13,18,20,28,36,37. En nuestra muestra, se utilizó la azatioprina en 2casos: en una paciente anciana en monoterapia por tener un mejor perfil de efectos adversos y en otro caso como segundo fármaco añadido a corticoides, en el que fue necesario asociar IGIV en la evolución por respuesta clínica insuficiente. En el caso del micofenolato, se empleó en un caso, aunque fue cambiado poco después a metotrexato por mala tolerancia. La paciente con una evolución lentamente progresiva fue tratada con rituximab ante la falta de respuesta a otros fármacos, sin lograr una mejoría clínica significativa. Por último, se ha propuesto el uso de plasmaféresis en casos graves con base en un potencial papel patogénico de los anticuerpos, sin haber llegado a un claro consenso para recomendar su uso17,28,38-40. En esta serie, el paciente con anticuerpos anti-HMGCR positivos tratado no mostró ningún beneficio tras 8sesiones.
El número de pacientes que alcanzaron una buena respuesta clínica fue algo mayor en el subgrupo con actividad espontánea en el estudio neurofisiológico frente al que no (75% versus 50%) y en el subgrupo con HLA tipo 1 positivo en la biopsia muscular (88,9% versus 60%).
En el seguimiento a los 6meses, a pesar de que más de la mitad de los pacientes llegaron a una resolución completa o lograron una mejoría significativa de los síntomas, 8pacientes seguían requiriendo la combinación de varios fármacos en el tratamiento. Un hito importante es la baja tasa de recaídas (12,5%), posiblemente relacionado con un mejor pronóstico de las formas asociadas a anticuerpos anti-HMGCR, altamente prevalentes en esta cohorte, frente a los anti-SRP20.
En cuanto al diagnóstico de cáncer, el cáncer asociado a miositis en MNIM es controvertido y se relaciona sobre todo con formas seronegativas de la enfermedad41. En nuestra muestra, por el contrario, el único caso de cáncer se dio en un caso de MNIM asociada a anti-HMGCR, lo que apoyaría el posible riesgo aumentado de cáncer en pacientes con formas de la enfermedad asociadas a estos anticuerpos14,30,42,43.
Este estudio tiene limitaciones, al tratarse de una serie de casos con una muestra de pequeño tamaño, tener un diseño retrospectivo y debido a la ausencia de escalas funcionales aplicadas de forma sistemática durante la evaluación y el seguimiento de los pacientes.
Más estudios son necesarios para determinar diferentes patrones de evolución en los distintos subgrupos de pacientes, lo que permitiría intentar predecir la respuesta clínica de forma precoz.
ConclusionesLa MNIM es la última entidad dentro de las MII en ser reconocida como un subgrupo diferente, cuyo diagnóstico ha aumentado en los últimos años. En nuestra muestra los únicos autoanticuerpos asociados a miositis que se detectaron fueron los anti-HMGCR, la mayoría de las veces en el contexto de tratamiento previo con estatinas. La determinación de estos autoanticuerpos y la biopsia muscular siguen siendo piezas clave para el adecuado diagnóstico precoz y para iniciar lo antes posible el tratamiento, que debe individualizarse en función de la respuesta a corticoides. Es necesario combinar varios fármacos para alcanzar una respuesta clínica. El uso de otras terapias, como plasmaféresis, es controvertido.
Aunque la mayoría de los pacientes presentaron un pronóstico favorable a los 6meses y la tasa de recaídas fue baja, es importante tener en cuenta que hay diferentes subgrupos de pacientes y que algunos presentan un curso clínico atípico con peor respuesta a inmunosupresores, además de que la posibilidad de un cáncer oculto debe tenerse en cuenta no solo en pacientes con formas seronegativas de la enfermedad.
FinanciaciónEsta investigación no recibió ninguna subvención específica de agencias de financiación en los sectores público, comercial ni sin fines de lucro.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

