



Ohtahara syndrome (OS, OMIM #308350, ORPHA1934) is an early-onset epileptic encephalopathy (EOEE) characterised by spasms, intractable seizures, suppression-burst pattern on the electroencephalogram, and severe psychomotor retardation. Mutations in STXBP1 – a gene that codes for syntaxin binding protein 1 and is involved in synaptic vesicle exocytosis – has been identified in most patients with OS.
Patient and resultsWe report the case of a 19-month-old child with OS who displays a previously unreported mutation in STXBP1 (c.1249+2T>C, G417AfsX7). This mutation is located in a donor splice site and eliminates exon 14, resulting in a truncated protein.
ConclusionThis previously unreported STXBP1 mutation in a subject with Ohtahara syndrome and non-lesional magnetic resonance imaging (MRI) broadens the mutational spectrum associated with this devastating epileptic syndrome.
El síndrome de Ohtahara (SO, OMIM #308350, ORPHA1934) es una encefalopatía epiléptica de inicio precoz (EEIP) caracterizada por espasmos, crisis epilépticas intratables, un trazado electroencefalográfico de brote-supresión y retraso psicomotor grave. En la mayoría de los pacientes con SO se han identificado mutaciones en el gen STXBP1, que codifica para la proteína de unión a sintaxina 1 y que está implicado en el mecanismo de exocitosis de las vesículas sinápticas.
Paciente y resultadosSe presenta el caso clínico de un varón de 19 meses de edad diagnosticado de SO en el que se ha identificado una mutación no descrita (c.1249+2T>C, G417AfsX7) en el gen STXBP1. La mutación está localizada en uno de los sitios donadores implicados en el procesamiento del ARNm del gen, lo que produce la pérdida del exón 14 y el posterior truncamiento de la proteína que codifica.
ConclusionesEsta nueva mutación en el gen STXBP1, identificada en un paciente sin lesión cerebral estructural subyacente, amplía el espectro mutacional asociado a este devastador síndrome epiléptico.
Ohtahara syndrome (OS, OMIM #308350, ORPHA1934) is an early infantile epileptic encephalopathy (EIEE) characterised by spasm, intractable epileptic seizures, suppression-burst pattern on EEG, and severe psychomotor impairment.1 The possibility of a genetic cause should be considered in those patients with no structural brain alterations or underlying metabolic dysfunctions. Mutations in the STXBP1 gene have been found in most patients with OS.2,3
STXBP1 codes for syntaxin 1-binding protein (STXBP1), which regulates synaptic vesicle fusion by attaching to soluble N-ethylmaleimide-sensitive factor attachment protein receptors (SNARE) proteins.4 The SNARE complex plays a major role in the fusion of synaptic vesicles to the presynaptic plasma membrane, which results in the release of neurotransmitters into the synaptic cleft.5
Our study describes a new mutation in one of the splice donor sites involved in mRNA processing of the STXBP1 gene in a patient with OS and no underlying brain lesions.
Patient and methodsOur patient was diagnosed with OS in the paediatric neurology department at Hospital Virgen de la Salud, Toledo, and subsequently referred to the neurology laboratory at the Fundación Jiménez Díaz Health Research Institute for a genetic study.
Clinical caseOur patient was a 19-month-old boy whose parents were healthy and nonconsanguineous and had no family history of epilepsy. During his first 15 days of life, he experienced tonic and focal clonic seizures, and oculogyric crises. At the age of 45 days, his EEG showed a suppression-burst pattern suggestive of OS. Physical examination revealed no dysmorphism and results from metabolic studies were normal. When the patient was 5 months old, he started to experience spasms. Treatment with vigabatrin and corticosteroids was effective, but the patient subsequently developed refractory complex partial seizures. He currently has severe psychomotor retardation: he is unable to talk or hold up his head and exhibits poor visual fixation and generalised hypotonia.
MethodsGenomic DNA was obtained from peripheral blood lymphocytes according to standardised procedures. We sequenced the STXBP1 gene in both directions in the amplified fragments from genomic DNA using polymerase chain reaction amplification, a sequencing kit (Life Technologies, Carlsbad, CA, USA), and an ABI 3130 DNA sequencer (Life Technologies) with primers specific for STXBP1. The analysis included exonic regions, exon–intron regions, and 5′ and 3′ regulatory regions in the sequence of STXBP1. The reference sequence used to analyse STXBP1 mRNA was NM_001032221.3 (NCBI reference sequence).
We screened for this novel variant in 165 healthy individuals.
cDNA was obtained by reverse transcription polymerase chain reaction from total mRNA using an oligo (dT) primer and ImProm-II Reverse Transcription System (Promega, Fitchburg, WI, USA). cDNA was subsequently amplified with gene-specific primers (5′-GAAGTCACCCGGTCTCTGAA-3′ [direct] and 5′-CACCGTGAGAGCTGGTAGGT-3′ [reverse]) to obtain a fragment including exons 11 through 16 of STXBP1.
The mutation was analysed with the Human Splicing Finder programme (http://www.umd.be/HSF/), a bioinformatics tool used to predict the effects of mutations in the areas involved in mRNA processing.6
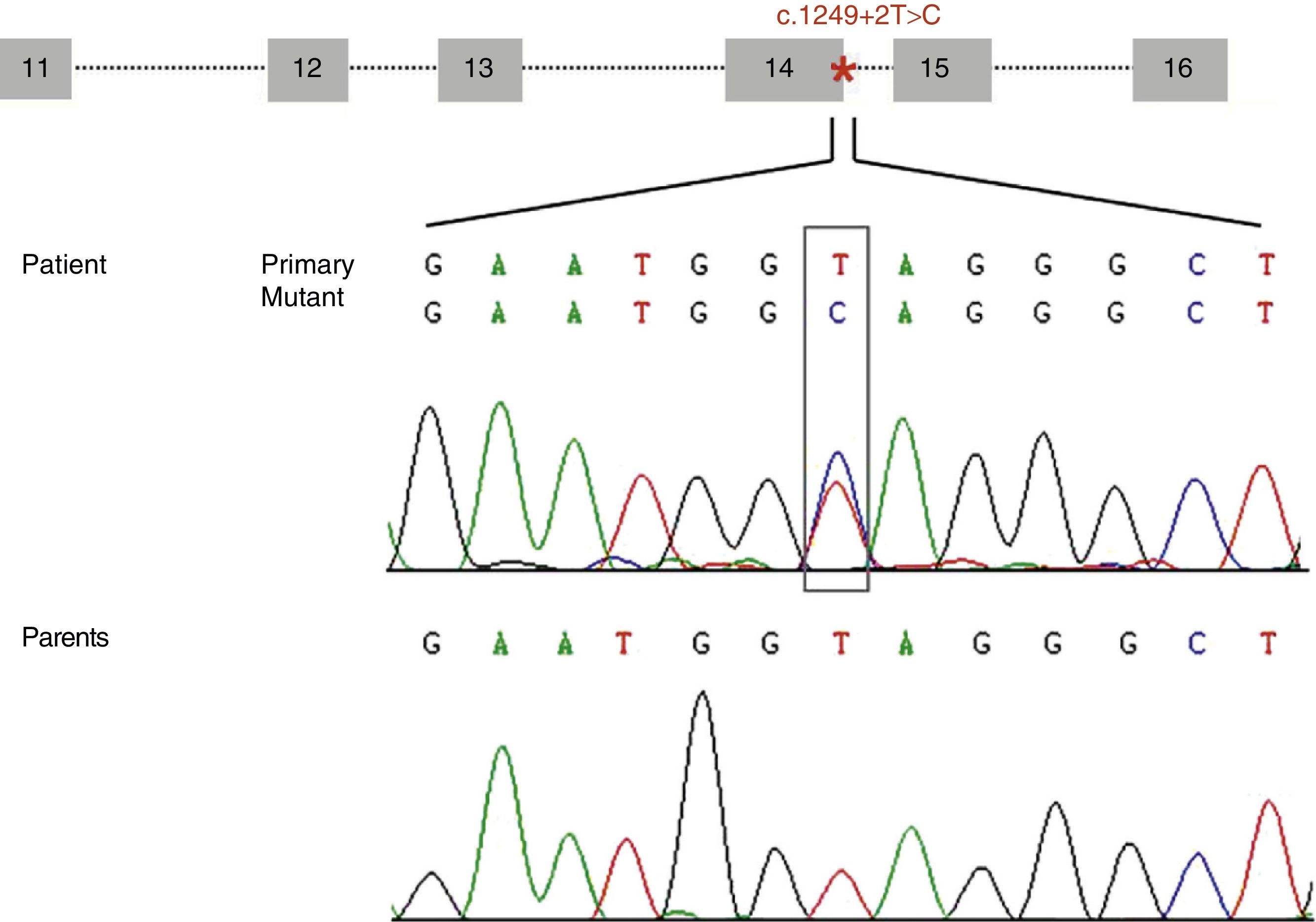
ResultsWe identified a novel heterozygous mutation of the STXBP1 gene (c.1249+2T>C) located at the splice donor site involved in mRNA processing at intron 14. This mutation was not found in our patient's parents (Fig. 1) or in any of the 165 controls we analysed. Analysis of this variant using the bioinformatics tool suggests that this mutation decreases the affinity of the mRNA processing machinery for the splice donor site with the mutation (ΔCV=−31.94). Activation of cryptic splice sites was not predicted.
 of STXBP1 containing the mutation c.1249+2T>C (located at intron 14 and marked with an asterisk). The electropherogram shows the mutation c.1249+2T>C identified in our patient and the corresponding sequence in each of his parents, which suggests that it is a de novo mutation.")
Diagram of the genomic structure of the fragment (exons 11 to 16) of STXBP1 containing the mutation c.1249+2T>C (located at intron 14 and marked with an asterisk). The electropherogram shows the mutation c.1249+2T>C identified in our patient and the corresponding sequence in each of his parents, which suggests that it is a de novo mutation.
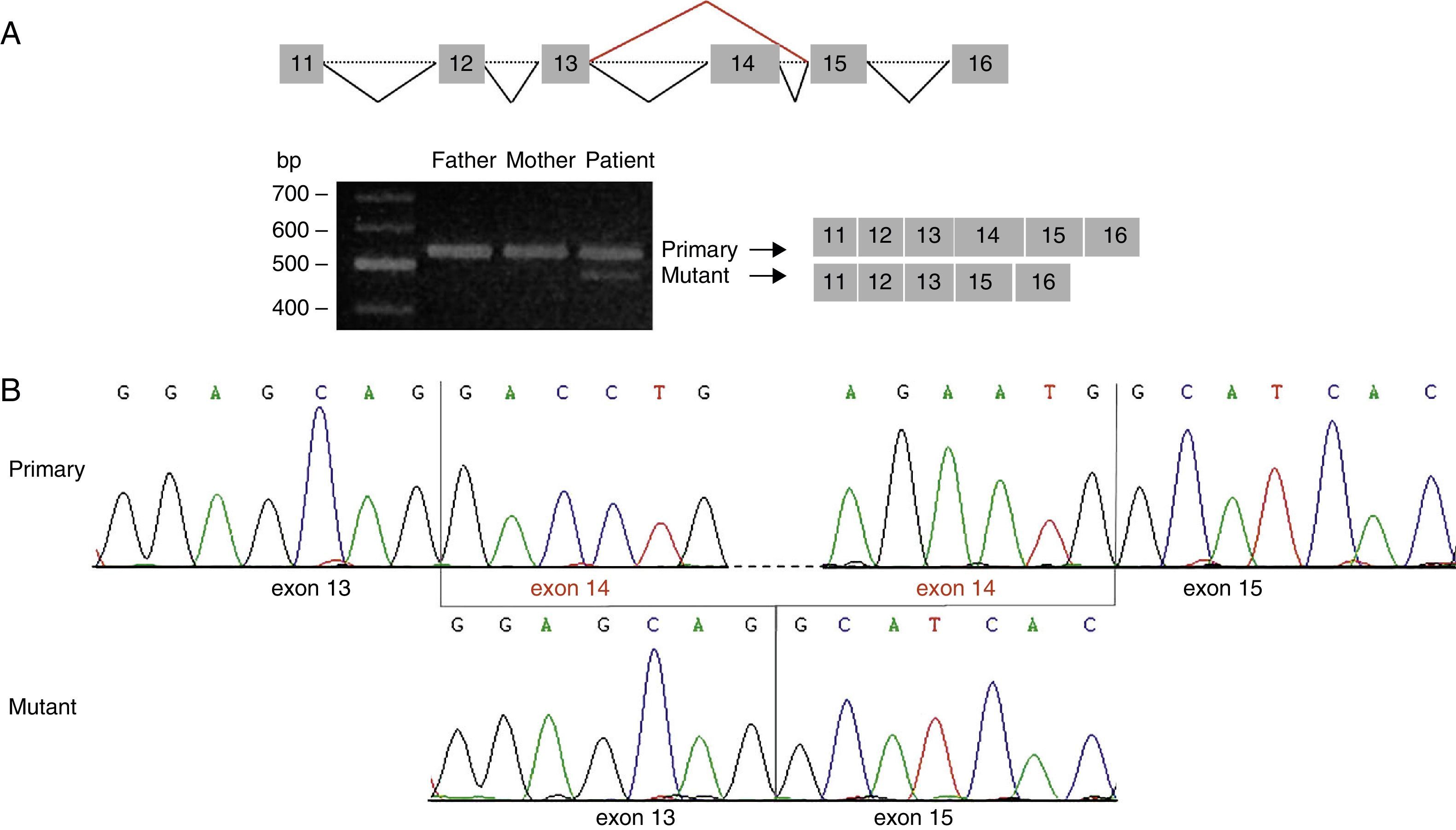
This prediction was confirmed by amplification of the cDNA region containing the identified variant. We detected a single band of 559bp (amplicon corresponding to the primary transcript) in the parents’ cDNA, but 2 bands in the patient's cDNA: a band of 559bp and another of 420bp (the amplicon corresponding to the mutant transcript) (Fig. 2A). Direct sequencing analysis revealed that the 420bp amplicon is lacking exon 14 and results in the creation of a new stop codon at position 424 of the protein sequence (G417AfsX7) (Fig. 2B).
 Amplification of the cDNA region corresponding to exons 11 to 16 of STXBP1 showed 2 different amplicon sizes in our patient: a band of 559bp (also seen in the patient")
Analysis of the mutation c.1249+2T>C by cDNA sequencing. (A) Amplification of the cDNA region corresponding to exons 11 to 16 of STXBP1 showed 2 different amplicon sizes in our patient: a band of 559bp (also seen in the patient's parents) and a band of 420bp, which corresponded to the amplicon of the mutant transcript. (B) Sequencing of the amplicon of 420bp confirms loss of exon 14 from STXBP1.
We identified a new mutation of STXBP1 in a patient diagnosed with OS who had no underlying structural brain lesions.
Since 2008, STXBP1 mutations have been reported in patients with OS, West syndrome, and EIEE, and in approximately 22% of the patients with non-lesional OS.2,3,7
The STXBP1 gene is located on chromosome 9q34.11, contains 20 exons, and codes for STXBP1. This protein regulates synaptic vesicle fusion and neurotransmitter release by binding to syntaxin-1A (STX1A), changing its conformation and regulating the SNARE complex.5
This novel mutation (c.1249+2T>C) eliminates the donor splice site involved in mRNA processing at intron 14. At the protein level, this mutation results in complete loss of domain 3b and part of domain 2 of STXBP1. Domains 1 and 3a form the central cavity providing the binding surface for syntaxin; this is an essential step in the formation of the SNARE complex and subsequent release of neurotransmitters.8 The identified mutation should therefore not affect STXBP1 binding to STX1A.
The literature describes mutations in the same functional domain as STXBP1,9 which points to the pathogenicity of this novel mutation. In addition, truncation of the Caenorhabditis elegans orthologue of STXBP1 downstream of position p.Y402 may impair synaptic vesicle fusion. The mutant transcript is likely to be degraded by nonsense-mediated mRNA decay (NMD), which results in haploinsufficiency of STXBP1, as reported in other studies describing similar mutations.10
This novel mutation in STXBP1, identified in a patient with non-lesional OS, broadens the spectrum of mutations associated with this devastating epilepsy syndrome.
Ethical standardsAll participants and/or their legal representatives signed informed consent forms approved by the ethics committee at Fundación Jiménez Díaz.
FundingThis study was funded by the Spanish Ministry of Economy and Competitiveness (SAF2010-18586 and EUI-EURC-2011-4325). Laura Ortega Moreno has received a research scholarship from Fundación Conchita Rábago de Jiménez Díaz.
Conflict of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Ortega-Moreno L, Giráldez BG, Verdú A, García-Campos O, Sánchez-Martín G, Serratosa JM, et al. Nueva mutación en el gen STXBP1 en un paciente con síndrome de Ohtahara no lesional. Neurología. 2016;31:523–527.
This study was presented in poster format in the section of Neurodegenerative Diseases at the European Human Genetics Conference held in Paris in 2013.

Neurología (English Edition) sigue las recomendaciones para la preparación, presentación y publicación de trabajos académicos en revistas biomédicas