



Opsoclonus-myoclonus-ataxia syndrome is a rare neuroinflammatory disorder with onset during childhood; aetiology may be paraneoplastic, para-infectious, or idiopathic. No biomarkers have yet been identified, and diagnosis is clinical. Better cognitive prognosis appears to be related to early onset of immunomodulatory therapy.
MethodsWe describe the epidemiological, clinical, therapeutic, and long-term prognostic characteristics of a cohort of 20 Spanish patients.
ResultsThe mean age of onset was 21 months (range, 2-59). Ataxia and opsoclonus were the most frequent symptoms both at disease onset and throughout disease progression. The mean time from onset to diagnosis was 1.1 months. Neuroblast lineage tumours were detected in 45% of patients; these were treated with surgical resection in 7 cases and chemotherapy in 2. Cerebrospinal fluid analysis revealed pleocytosis in 4 cases (25%) and neither antineuronal antibodies nor oligoclonal bands were detected in any patient. Immunomodulatory drugs were used in all cases. Nine patients started combined immunomodulatory treatment at the time of diagnosis, and 5 patients after a mean of 2.2 months. In the long term, 6 of the 10 patients followed up for more than 5 years presented mild or moderate cognitive sequelae. Four patients presented relapses, generally coinciding with the decrease of corticosteroid doses.
ConclusionsEarly initiation of immunotherapy, as well as triple combination therapy, where needed, was associated with a lower frequency of cognitive impairment 2 years after onset.
El síndrome opsoclono-mioclono-ataxia es un raro trastorno de inicio pediátrico; de base neuroinflamatoria y origen paraneoplásico, parainfeccioso o idiopático. Actualmente no hay biomarcadores, siendo el diagnóstico clínico. El pronóstico cognitivo parece estar relacionado con el inicio temprano de la terapia inmunomoduladora.
MétodoSe describen las características epidemiológicas, clínicas, terapéuticas y pronósticas a largo plazo de una cohorte de 20 pacientes españoles.
ResultadosLa edad media de debut fue de 21 meses (2−59 meses). La ataxia y el opsoclonus fueron los síntomas de inicio más frecuentes y predominantes en la evolución. El tiempo medio desde los primeros síntomas hasta el diagnóstico fue de 1,1 mes. Un tumor de extirpe neuroblástica fue detectado en el 45 %, realizándose resección quirúrgica en 7 y quimioterapia en 2. En el estudio de líquido cefalorraquídeo se constató pleocitosis en 4 (25%), con negatividad de anticuerpos antineuronales y bandas oligoclonales en todos los casos estudiados. En el 100% se emplearon fármacos inmunomoduladores. En 9 pacientes el tratamiento combinado inmunomodulador se inició desde el momento del diagnóstico, y en 5 el tiempo medio de implementación fue de 2,2 meses. A largo plazo, 6 de 10 pacientes con seguimiento superior a 5 años presentaban secuelas cognitivas leves o moderadas. Cuatro pacientes presentaron recaídas, generalmente coincidiendo con el descenso de la corticoterapia.
ConclusionesEl inicio precoz de la inmunoterapia, así como de la triple terapia en los casos que lo precisaron, se relacionó con una menor frecuencia de afectación cognitiva a los 2 años del debut.
Opsoclonus-myoclonus-ataxia syndrome (OMAS) is a probably neuroimmunological disease defined clinically according to the presence of motor symptoms; the cardinal symptoms are ataxia, myoclonus (muscle jerks of central origin), and opsoclonus (erratic, multidirectional eye movements). This triad is generally accompanied by irritability and/or insomnia, in different combinations.1 These symptoms are sometimes associated with neural crest tumours (usually neuroblastoma), which constitutes another defining element in the disease.1,2 Some authors have attempted to simplify the diagnosis of OMAS, with diagnosis established according to the presence of 3 of the following signs and symptoms: opsoclonus or ocular flutter; myoclonus and/or ataxia; neuroblastoma; and mood swings and/or sleep disorders, frequently associated with irritability.1,2 Incidence is estimated at 0.18 cases per million population3, and 0.27-0.40 cases per million children4.
In up to 50% of cases, OMAS is associated with neural crest tumours, including neuroblastoma, ganglioneuroblastoma, and ganglioneuroma. Some authors suggest that this percentage may be far greater, but that a humoural or cellular immune response destroys the tumour mass before it can be detected.5 In addition to tumour processes, the disease displays a temporal association with various infections, including those caused by Epstein-Barr virus,6Mycoplasma pneumoniae,7,8 West Nile virus,9 hepatitis C virus,10 C3 adenovirus,11 and rotavirus.12 However, it is difficult in many cases to establish the true cause, with aetiology being considered idiopathic. In all cases of OMAS, the cardinal symptoms appear to be caused by an immune-mediated mechanism affecting not only the brainstem and cerebellum, but also supratentorial structures.13
Many cases have been reported of patients with neurocognitive and psychiatric sequelae, although prognosis has improved in recent years with the early administration of combination therapies.14 Despite this, controversy remains with regard to the impact of delayed diagnosis or treatment on these cognitive and behavioural aspects.15–17
We present a series of 20 Spanish paediatric patients with OMAS, describing the epidemiological, clinical, aetiological, treatment, and long-term prognostic characteristics.
Material and methodsPatientsData were gathered on patients younger than 18 years diagnosed with OMAS at hospitals in 6 Spanish cities (Madrid, Pamplona, Valladolid, San Sebastián, Vitoria, and Granada) between 2006 and 2018. Information was collected through a standardised questionnaire.
We included patients with a diagnosis of OMAS who presented 3 of the following signs: 1) opsoclonus or ocular flutter; 2) neuroblastoma; 3) myoclonus and/or ataxia; 4) mood swings and/or sleep disorders, often associated with pronounced irritability.
Clinical dataWe analysed information on sex, age of onset, signs and symptoms of OMAS, age at diagnosis, symptom chronology and progression, delay in diagnosis and treatment onset; physical examination using specific rating scales (Pranzatelli and Mitchell-Pike scales),15,18 complementary testing (infection, immunological, and tumour screens), treatments received and form of implementation, and long-term progression in terms of cognition, behaviour, motor involvement, and sleep disorders.
Statistical analysisThe homogeneity of demographic and clinical variables was analysed during the study. Data are presented as mean, median, range, and standard deviation for quantitative variables, and absolute and relative frequencies for qualitative variables. Due to the small sample size, quantitative variables were analysed with nonparametric statistics (Spearman rho). Qualitative variables were analysed for homogeneity with the chi-square test or the Fisher exact test, as appropriate. Values of P < .05 were considered statistically significant. Statistical analysis was performed with the SPSS software (version 17.0; IBM Inc.).
ResultsClinical and epidemiological characteristicsWe analysed data from a total of 20 patients with OMAS; epidemiological characteristics and form of presentation are summarised in Table 1. Follow-up time ranged from 3 months (last patient diagnosed) to 12 years, with a median of 7 years.
Clinical characteristics at onset in 20 patients with opsoclonus-myoclonus-ataxia syndrome.
| Clinical characteristics | n | % | Mean (SD) | Range | Comments |
|---|---|---|---|---|---|
| Female sex | 11 | 55 | |||
| Mean age at diagnosis (months) | 21 (11.8) | 2-59 months.90% aged 14-30 months.10% aged ≤ 6 months | Similar in girls and boys (21 vs 21.3 months) | ||
| Diagnostic delay (months) | 1.1 (1.78) | ||||
| Prodromal symptomsTotalParainfectious cases | 10 | 50 | 5 respiratory infections (URTI/AOM/bronchitis), 2 AGE, 2 fever without focus, 1 urticarial rash | ||
| 3/10 | 33 | Mycoplasma pneumoniae IgM, rhinovirus (nasoph.), RSV (nasoph.) | |||
| Initial manifestationAtaxiaOpsoclonus | 146 | 7030 | |||
| Cardinal symptomsAtaxiaIrritabilityOpsoclonusSleep disorderMyoclonusSpeech disorder | 19181716136 | 959085806530 | 7/18 (39%) required pharmaceutical treatment10/16 (62.5%) required pharmaceutical treatment (melatonin) |
Percentages shown as a proportion of the total sample. AGE: acute gastroenteritis; AOM: acute otitis media; IgM: immunoglobulin M; nasoph.: nasopharyngeal; RSV: respiratory syncytial virus; SD: standard deviation; URTI: upper respiratory tract infection; n: It is the number of patients.
Not all patients presented the cardinal symptoms of OMAS (opsoclonus, myoclonus, ataxia, irritability, and insomnia). The earliest symptoms were ataxia and opsoclonus, which were also the most frequent symptoms over the progression of the disease, alongside irritability. Among patients with onset after 2 years of age (n = 6), one-third did not present opsoclonus, whereas this symptom was present in all but one of the 14 patients with earlier onset (7%). In 3 patients not presenting clear myoclonus, the initial symptom was tremor. At admission, only 5 patients (25%) met the clinical criteria for diagnosis. OMAS duration (from onset to resolution of all symptoms) was acute (< 3 months) in 4 patients, subacute (3-12 months) in 5, and chronic (> 12 months) in 8. In 3 patients, the precise time of clinical resolution was not known.
At onset, only 6 patients (30%) underwent systematic examination with the Mitchell-Pike scale, and 10 (50%) with the Pranzatelli scale; long-term follow-up (6, 12, and 24 months) with these scales was conducted for 6 (30%) and 7 (35%) patients, respectively. At onset, the mean (SD) scores on these scales were 11.5 (1.1) for the Mitchell-Pike scale and 21.8 (2.52) for the Pranzatelli scale. Among patients tested with the Pranzatelli scale, only one patient presented mild involvement (defined as a score between 0 and 12), with 5 presenting moderate involvement (13-24) and 4 severe involvement (25-36).
Eight patients (40%) underwent neuropsychological assessment at diagnosis. Among those younger than 2 years at symptom onset, this assessment included the Battelle Developmental Inventory; assessment of older patients included the Wechsler intelligence scale (Primary and Preschool [WPPSI] and Children [WISC] versions, according to age). Some patients also completed other tests at some point over the progression of the disease (McCarthy Scales of Children’s Abilities [MSCA], Kaufman Assessment Battery for Children [K-ABC], Child Neuropsychological Maturity Questionnaire [CUMANIN], and Conners Continuous Performance Test [CPT] in patients with attention difficulties). Neuropsychological assessment conducted at diagnosis identified alterations in 5 patients (62%): 3 with mild global developmental delay (developmental quotient [Battelle Developmental Inventory] of 76-86), one of whom also had behavioural and attentional alterations; one with motor and attentional alterations; and one with language and global learning delay. During follow-up, and excluding the patient with a progression time below one year, only 6 patients (30%) underwent an additional cognitive/behavioural assessment.
Tumour characterisationNine patients (45%) presented neuroblastic tumours (tumour characteristics and therapeutic management are shown in Tables 2 and 3). In 2 cases, the tumour was detected prior to diagnosis of OMAS, which developed between 8 and 24 months later, with no recurrence of the tumour. Mean age of tumour onset was slightly lower for neuroblastoma than for ganglioneuroblastoma. There were no delays in the aetiological diagnosis of the tumour, which was established in the first screening study, including analysis of urine catecholamines, imaging studies, and bone scintigraphy with metaiodobenzylguanidine (MIBG). The imaging studies conducted included various combinations of the following: chest radiography, abdominal ultrasound, and CT and/or MRI of the targeted regions (if a suspicious mass had been detected in a previous test) or the whole body. The latter studies detected the tumour in all cases, even those not initially detected with chest radiography or abdominal ultrasound and in patients with negative MIBG and catecholamine study results.
Characterisation of tumours in 9 patients with paraneoplastic opsoclonus-myoclonus-ataxia syndrome.
| Tumour characteristics | n | % | Comments | |
|---|---|---|---|---|
| Type of tumour | Neuroblastoma | 7 | 78 | Mean age at tumour diagnosis: 14.7 months (range, 5-27) |
| Ganglioneuroblastoma | 2 | 22 | Aged 20 and 30 months at diagnosis, respectively | |
| Localisation | Adrenal or abdominal/paraspinal | 6 | 67 | |
| Posterior mediastinum | 1 | 11 | ||
| Paravertebral (not specified) | 2 | 22 | ||
| Complementary tests | Urine catecholamines | 9 | 100 | Abnormal in 3/9 (33%) |
| MIBG scintigraphy | 8 | 89 | Pathological uptake in the tumour area in 3/8 (37.5%) | |
| Targeted/whole-body MRI/CT | 9 | 100 | Tumour mass detected in 9/9 (100%), even those with normal chest RX and abdominal echography results with or without normal urine catecholamine and MIBG results | |
| Treatment | Complete surgical resection | 7 | 78 | 2 with OMAS onset 8 and 24 months later with no evidence of recurrence |
| Chemotherapy | 2 | 22 | One isolated due to inoperability, the other after surgery |
Percentages shown as proportion of all cases of paraneoplastic OMAS. CT: computed tomography; MIBG: metaiodobenzylguanidine; MRI: magnetic resonance imaging; OMAS: opsoclonus-myoclonus-ataxia syndrome; RX: radiography.
Clinical characteristics at onset and over the course of the disease in different groups (paraneoplastic cases and idiopathic/cryptogenic cases with/without associated infection).
| Variable | Paraneoplastic | Idiopathic/cryptogenic | |
|---|---|---|---|
| Presumably parainfectious | Without known history of infection | ||
| No. patients | 9 | 7 | 4 |
| Mean age of onset | 20.22 | 26.42 | 14 |
| Prodromal symptoms | 3 (33%) | 7 (100%) | 0 |
| Time until 3 of 4 diagnostic criteria met (months) | 3.12 | 0.63 | 1.9 |
| Second-line treatment | 3 (33%) | 1 (14%) | 3 (75%) |
| Currently asymptomatic | 6 (66%) | 5 (55.5%) | 1 (25%) |
| Current symptom persistence | 2 (22.2%) | 0 | 3 (75%) |
| Patients lost to follow-up | 1 | 2 | 0 |
| Mean time to complete resolution (months)a | 19.1 | 3.4 | 30 |
| Cognitive and/or behavioural sequelae at 2 yearsb | 4 | 1/6(b 1) | 2/2(b 2) |
| Sleep disorder at 2 years | 1 | 0/6(b 1) | 1/2(b 2) |
| Other sequelae at 2 years | Residual ataxia (1) | 0/6 | Mild motor coordination problems (2/4) |
In 7 patients presenting clear signs of infection in the weeks prior to onset of OMAS (5 respiratory and 2 gastrointestinal infections), and for whom no related aetiology was established at onset or subsequently, aetiology was assumed to be parainfectious (Table 3). Strikingly, clinical symptoms resolved earlier in this patient group, with less need for second-line treatments.
NeuroinflammationSixteen patients (80%) underwent lumbar puncture, which revealed pleocytosis (leukocyte count ≥ 4 cells/mm3) in 4 cases (25%). CSF glucose level was normal in all patients, with one presenting an elevated protein level (85 μg/L; normal range, 15-45). A study of the blood-brain barrier, including oligoclonal bands, was conducted in 8 cases (50%); a band in the beta-globulin spectrum was detected in one patient with presumably parainfectious aetiology due to Mycoplasma pneumoniae infection. In 9 of 17 patients (53%), studies were conducted to detect antineuronal antibodies in the serum, CSF, or both; all tests yielded negative results.
Immunomodulatory treatmentAll patients received some form of immunomodulatory treatment. In most cases, treatment was started at the time of diagnosis, with the mean time from symptom onset to immunotherapy onset (1.1 months; SD: 1.78) being equivalent to the diagnostic delay. The mean delay from definitive diagnosis to treatment onset was 0.3 months (range: 0 days to 1.5 months). Immunomodulatory treatment included monotherapy with corticosteroids or ACTH (no patient received immunoglobulins in isolation), with 14 patients (70%) receiving combination therapy: 8 received second-line drugs (rituximab in 6 and cyclophosphamide in 2), which were added to previous bitherapy with immunoglobulins and corticosteroids in 7 cases, and to monotherapy with corticosteroids in one.
In 9 children, immunomodulatory combination therapy was started from the time of diagnosis; in the remaining 5, the mean time to onset of immunomodulatory combination treatment was 2.2 months (range, 15 days to 4 months). Six patients needed a combination of ≥ 3 treatments to control symptoms. The mean time from onset of mono- or bitherapy to the introduction of a second-line drug was 8 months (range, 3-26).
Patients received a mean of 14 cycles of immunoglobulins (range, 1-36). Six patients received rituximab, with 2 needing an additional cycle of 4 doses of 375 mg/m2. The 2 patients treated with cyclophosphamide received the drug on a monthly basis (3 and 8 doses, respectively). Data on the overall duration of immunomodulatory treatment is unavailable for 4 patients; another 4 continue receiving this therapy at present (range, 3-27 months). In the remaining 12 patients, mean duration was 17.6 months (range, 1-36).
Progression, relapses, and sequelaeOf the 8 patients with chronic progression (> 12 months from OMAS onset), 4 presented symptom resolution at 17-55 months of follow-up, with no relapses; in the remaining 4, symptoms persist, with duration ranging from 13 months to 11 years. All 4 continue to present ataxia, with 2 (with shorter progression times, 12 and 24 months) presenting it continuously; one of these also presents irritability. The other 2 (with progression times of approximately 10 years) present mild, intermittent ataxia (in the context of fever or infection). Overall, mean time to symptom resolution in the 12 asymptomatic patients was 11.72 months (range, 0.5-55), with differences between symptoms in the time to resolution (Table 4).
Progression of the cardinal symptoms.
| Symptom | Cases with complete resolution (n [%]) | Time to complete resolution (mean [SD]; range) | Comments |
|---|---|---|---|
| Opsoclonus | 17 (100%) | 3.7 (3.4) months (0.05-10) | |
| Myoclonus | 13 (100%) | 4.7 (3.7) months | |
| Ataxia | 15 (78.9%) | 9 (7.9) months (median: 5) | Symptom taking longest to resolve |
| Irritability | 15/18 (83%) | Median: 5 months | One patient developed a behavioural disorder. |
Percentage of all patients with each symptom in whom the symptom resolved. SD: standard deviation.
Although cognitive and behavioural sequelae were the most common (Table 5), it was only possible to determine the initial symptom and its progression in the 6 patients undergoing baseline neuropsychological assessment and subsequent follow-up assessments. In 2 of these patients, who initially presented mild global involvement (DQ of 73 and 86, respectively), we observed an improvement to the point that they achieved normal values (intelligence quotients of 94 and 106, respectively, on the Wechsler intelligence scale at 2 years after onset). The 2 patients presenting attentional difficulties in the initial evaluation continued to present alterations to executive function in follow-up studies (attention-deficit/hyperactivity disorder [ADHD]) (follow-up times of 5 and 10 years, respectively). One of these patients underwent follow-up assessments after starting treatment with stimulants, achieving normal values for specific parameters. Two patients developed a specific learning disorder of reading and writing: one presented initially normal neuropsychological assessment results, and the other had previously presented language delay, but with favourable progression at 10 years of follow-up.
Sequelae.
| Follow-up time | N | No. seq. | Diagnosis (sequela) |
|---|---|---|---|
| > 10 years | 3 | 3 | Reading/writing difficultiesReading/writing difficulties + ADHDHyperactive type ADHD |
| 5-10 years | 7 | 4 | ADHD (2 cases)Reading/writing difficultiesGlobal poor motor coordination with mild residual ataxia |
| 2-5 years | 7 | 2 | Persistent irritabilityLanguage delay |
| < 2 years | 3 | Not assessed due to short progression time; 2 patients with persistent acute cardinal symptoms. |
N: number of patients followed up for the indicated time period; no. seq.: number of patients presenting sequelae; ADHD: attention-deficit/hyperactivity disorder.
Four patients (25%) presented clear relapses. Three had OMAS associated with neuroblastoma, fully resected at symptom onset, with subsequent negative tumour screening test results at 7.5, 9, and 9.5 years of follow-up, respectively. Two of the 4 had been treated with monotherapy and the other 2 with combination therapy. One, who had received second-line treatment, relapsed after 2 years without symptoms. Immunotherapy was restarted with immunoglobulins plus a second cycle of rituximab, with symptoms resolving several months later; no further relapses were recorded. The other patient presented 2 relapses, both following the discontinuation of corticotherapy (prednisone), which he was receiving in combination with immunoglobulins; the relapse was treated with a cycle of rituximab plus azathioprine. Among the patients treated with monotherapy, one presented a relapse after the beginning of ACTH withdrawal, 4 months after symptom onset, having initially presented complete treatment response. The other (the only patient with idiopathic OMAS) relapsed 5 months after the discontinuation of corticosteroids (prednisone); treatment was restarted at the full dose, achieving a clinical response.
Potential prognostic factorsRegarding the possible prognostic repercussions of early onset and severity at onset, we observed a statistically significant association (Spearman rho of 1; P = .01) between greater initial clinical severity according to the Mitchell-Pike scale and time to complete symptom resolution. No such association was observed for the Pranzatelli scale. It should be noted that baseline Mitchell-Pike scale scores were only available for 6 patients, with data from the Pranzatelli scale being available for 10 (data from both scales were available for 3 patients), which may have influenced the findings. We also observed a certain trend towards longer symptom duration in patients with early onset, although this association was not statistically significant.
Early onset was defined as the appearance of symptoms before 18 months of age, as this corresponds to a stage of neurological development at which the majority of healthy children are able to walk independently and to use expressive language. No significant difference in the frequency of cognitive involvement in the 2 years after onset was observed between patients with early and late onset (16% vs 36%; P = .6). Two patients with follow-up times shorter than 2 years were excluded from this analysis. We did not analyse the association with cognitive involvement at 5 years, as sufficient follow-up data were only available for 10 patients. The early-onset group more frequently needed second-line drugs (62.5% vs 25%), although this difference was not significant (P = .16).
Regarding the importance of early treatment onset, the time from symptom onset to onset of immunotherapy showed no statistically significant correlation with clinical scale scores (Mitchell-Pike and Pranzatelli scales) at 6, 12, and 24 months. However, data from either of these scales were only available for 11 patients (and data from both scales for only 6 patients), which may have influenced this analysis. Neither did we observe a correlation between treatment delay and time to symptom resolution (Pearson r of 0.25; P = .42). Early onset of any form of immunomodulatory treatment (within 2 months after symptom onset) was associated with lower incidence of cognitive involvement at 2 years (13% vs 50%); while not statistically significant, this difference is clinically relevant.
In patients requiring second-line drugs, delayed onset of this treatment was associated with delayed resolution of OMAS symptoms (Spearman rho of 0.95; P = .05). The frequency of cognitive involvement showed no significant differences between patients who did and who did not receive second-line treatments (33% vs 27%). However, we did observe a relationship with early intensification of treatment: of the 8 patients treated with a second-line drug, 4 received the first dose in the first 6 months of immunotherapy (2 received it at 3 months), and 3 received the first dose beyond 6 months (2 at 10 months and one at 26). Six patients were previously receiving bitherapy (steroids and immunoglobulins); thus, the addition of the second-line drug constituted the beginning of tritherapy. All patients starting tritherapy in the first 6 months of treatment (3/3) were cognitively normal at 2 years after OMAS onset, whereas all 3 of those starting this treatment after the sixth month presented cognitive alterations at 2 years.
DiscussionTo our knowledge, this is the largest Spanish cohort to date of patients with OMAS and medium- to long-term follow-up data. This enables us to make certain epidemiological, clinical, and prognostic observations about the management of this rare disease in our setting that may be useful for current and future clinical practice. It should be noted that the small patient sample, due to the low prevalence of the disease, continues to represent a limitation for establishing robust conclusions.
Clinical and epidemiological characteristicsOur patient cohort showed a slight female predominance (55% of patients), very similarly to the findings of other cohorts, which report that 52 %-65 % of patients were girls.1,4,19,20 The apparent status of female sex as a risk factor, as occurs in other autoimmune/autoinflammatory diseases, requires further study. Onset most frequently occurred in preschool ages (14-30 months), with 2 patients (10%) presenting onset before 6 months of age.
The mean diagnostic delay (1.1 months) was very similar to the most recent figure reported by Pranzatelli et al.1 (1.2 months), and greater than those reported by Singhi et al.21 (2 months; range, 2 days to 12 months) and Huddar et al.22 (3.8 ± 5 months; range, 15 days to 16 months). This may be a key factor in starting early treatment and performing tumour screening, and may even influence long-term prognosis.15,16
Presence of ataxia (the most frequent initial manifestation1,23) as the sole symptom at onset hinders and delays diagnosis due to the need to consider acute postinfectious ataxia as the main differential diagnosis.21 However, this condition less frequently affects infants and often presents with prodromal signs and skin rash, mainly due to association with varicella zoster virus infection.23,24 The appearance of other behavioural symptoms, such as irritability and/or marked insomnia together with persistent ataxia, should raise suspicion of OMAS; this was the case in 2 of the 6 patients older than 2 years in our study, both of whom presented neuroblastoma. Some patients without myoclonus presented tremor. According to previous reports, mild myoclonus can have a tremulous appearance that can be mistaken for tremor; however, myoclonic jerks are usually more intense and are more clearly action-induced.1
In the clinical evaluation and follow-up of patients, it is desirable to use specific scales enabling stratification of severity, more objective evaluation of treatment response, recording of relapses, and comparisons and potential associations between clinical severity and prognostic variables. In this sense, the low percentage of patients who were systematically evaluated with these scales constitutes a limitation of our study. Similarly, despite the high prevalence of cognitive involvement and its potential repercussions, only 30% of patients (6/20) underwent neuropsychological evaluation. This may be due to the difficulty that many Spanish hospitals face in accessing paediatric neuropsychology teams.
Tumour characterisationThe frequency, tumour lineage, and the influence of age (greater predominance of ganglioneuroblastoma in older patients) observed in paraneoplastic cases were similar to the results of previous international series.1 As is also reported by other authors,25–28 the prognosis of the oncological disease was favourable, with all 9 patients presenting a complete recovery. All tumours were detected in the first screening study. However, as some authors speculate that tumours may regress spontaneously or may be too small for detection, the designation of the remaining patients as “non-paraneoplastic” may not be accurate in all cases.19,27
Despite the high sensitivity of MIBG for diagnosing neuroblastoma, with previous studies reporting figures of up to 95%,29 CT and MRI were more sensitive in our study; other researchers have also reported similar findings.21,23,30,31 The lower sensitivity of MIBG has been attributed to the low degree of malignancy/aggressiveness of the tumour in question.21,22,30,31 Biasotti et al.29 found an association between negative MIBG findings and normal urine catecholamine levels, which they attribute to the existence of non-secreting neuroblastomas (approximately 5%). As the tumours associated with OMAS are often low-grade, diagnosis based on tests of metabolic activity (such as MIBG and urine catecholamine metabolite profile) may not be sufficiently sensitive if used in isolation; therefore, it is essential also to conduct protocolised imaging studies.30 Furthermore, as a result of their smaller size, they may not be detected in simple radiography or ultrasound studies, giving rise to a need for more thorough techniques with multiple slices, such as CT or MRI.16 In the light of these considerations, whole-body CT, or preferably (due to the lower level of radiation) whole-body MRI, may be the most cost-effective diagnostic tool in these patients. It should be noted that some patients with paraneoplastic OMAS presented previous infectious symptoms, so paraneoplastic aetiology could not be ruled out.6,23
Characterisation of idiopathic/cryptogenic casesIn our series, “presumably infectious” aetiology was associated with better prognosis in terms of earlier symptom resolution, lower frequency of second-line treatment, and a lower percentage of sequelae at 2 years (Table 3).6,23
Characterisation of immunological statusOligoclonal bands constitute a possible manifestation of the pathogenic role of B cells.32 They were only determined in 50% of cases. A band was detected in a single patient with neuroblastoma; this is compatible with the findings of Pang et al.3 Our series did not display the potential relationship between positivity for oligoclonal bands and greater clinical severity, as was recently suggested by Pranzatelli.33Unlike other research groups, we identified no antineuronal antibodies.22,33–36
Progression, relapses, and sequelaeThe mean time to complete symptom resolution (11.72 months) was similar to that reported by other authors, such as Huddar et al.22 (12 months). Treatment was maintained for a mean of 17.6 months in our sample; this value is somewhat lower than those reported by other authors, including Huddar et al.22 (22.3 ± 20 months; range, 3 months to 5 years), although the latter study reported a relapse rate of 43% (6/14), higher than our own (20%). Therefore, other factors may have had an influence, such as the longer diagnostic and therapeutic delays or, in the case of our study, the lack of sufficiently long follow-up periods for some patients and the more intense, earlier immunotherapy used in recent years.
While no time period has been clearly established, relapses are usually defined as the reappearance and persistence of OMAS symptoms for at least 48-72 hours, while pseudo-relapses are defined as transient symptom exacerbation occurring in certain situations, such as sedation or anaesthesia, without symptom progression.37 Thus, the clinical course of OMAS may be monophasic or multiphasic30; without long follow-up periods, we cannot reliably establish which patients will present persisting remission and which will relapse.30
In our series, 11 patients presented a monophasic course, with no relapses during follow-up (range, 2-7.5 years). Four (20%) presented clearly relapsing-remitting courses, with 3 presenting a single relapse and one presenting 2. Three relapses were associated with withdrawal of corticotherapy, the most frequently reported trigger factor, followed by infection.38,39 One patient presented a relapse 2 years after the withdrawal of immunotherapy. In this case, the long period without symptoms or treatment supports the need for long follow-up in order to characterise the course of the disease, and demonstrates the heterogeneity of relapse patterns and the progression time at which they can occur.30 In this regard, cases have even been reported of relapses during adulthood.40 With respect to the previous immunotherapy administered, relapses were more frequent in patients treated in monotherapy (2/6) than in those who received polytherapy (2/14); this supports the use of combination therapy, as has been the recent trend.37 We should highlight the fact that 2 of the 4 patients with relapses presented cognitive sequelae; minimising these symptoms is one of the objectives of the new therapeutic approaches.
Two patients who initially presented remission occasionally display minor symptoms (nonspecific eye movements or mild ataxia, coinciding with infection); these symptoms are self-limiting without changes to treatment. These exacerbations probably best fit the concept of pseudo-relapse. Their appearance may reflect incomplete control of the disease, with an increased risk of true relapses,37,39 giving rise to a need for long-term follow-up.
The long-term progression of patients with OMAS is mainly characterised by neuropsychological sequelae, behaviour disorders, and learning difficulties, whereas the initial neurological symptoms tend to improve with immunotherapy32 or even spontaneously. In our series, the most frequent sequelae were ADHD and learning disorders affecting reading and writing; we did not observe a pattern of progressive deterioration with age, as reported by Mitchell et al.,41 who propose the possibility of progressive encephalopathy rather than a time-limited insult.
Potential prognostic factorsDespite some reports of spontaneous symptom resolution,3,21 immunomodulatory therapy is essential. Recent years have seen a shift in treatment approaches, with early intensification of treatment, bitherapy (generally with steroids and immunoglobulins) from treatment onset, and the early addition of a second-line drug if treatment response is unsatisfactory42. Some authors even propose starting with tritherapy.14 The aim of optimising treatment (and therefore improving the control of the underlying immunopathological process) is not only to control the acute symptoms, but also to minimise the permanent cognitive effects.
While some patients develop normally after OMAS, the majority present certain cognitive or neuropsychological deficits (involving attention, memory, visuomotor skills, and working memory) in the long term, generally with normal or nearly normal motor outcomes.23 Other series report some neuropsychological deficit in 80% of patients,43 although it is important when interpreting this information to be aware of the retrospective nature of most of these series and the diverse range of treatments used. Such factors as younger age at diagnosis, delays in implementing immunosuppressive treatment, greater clinical severity at onset, and relapses after reducing corticosteroid doses (and the consequent delay in complete resolution) have been suggested as potential predictive factors of poor prognosis.20,23
In our series, patients with early onset (before 18 months of age) and later onset presented no significant differences in the prevalence of cognitive involvement at 2 years after onset.
However, despite the lack of a statistically significant association between early onset of immunotherapy and better prognosis, we did observe an association with lower prevalence of cognitive sequelae at 2 years, which we consider to be clinically relevant. Similarly, while no differences in cognitive involvement were observed between patients who did and did not require second-line treatments, all patients starting tritherapy in the first 6 months of treatment presented normal cognition at 2 years, unlike those who started tritherapy later. Based on this finding, we consider it important to start treatment early and to consider tritherapy from the first months of treatment.
In our series, the time from symptom onset to onset of immunotherapy was approximately 1.1 months, with a mean delay from definitive diagnosis to treatment onset approaching 0.3 months. In any case, we may assert that today, in our setting, there is a tendency to start immunotherapy earlier, with generally shorter delays than those reported in such other series as that by Huddar et al.22 (mean time from diagnosis to immunotherapy onset of 6.1 [SD: 7.5] months; range, 2-24).
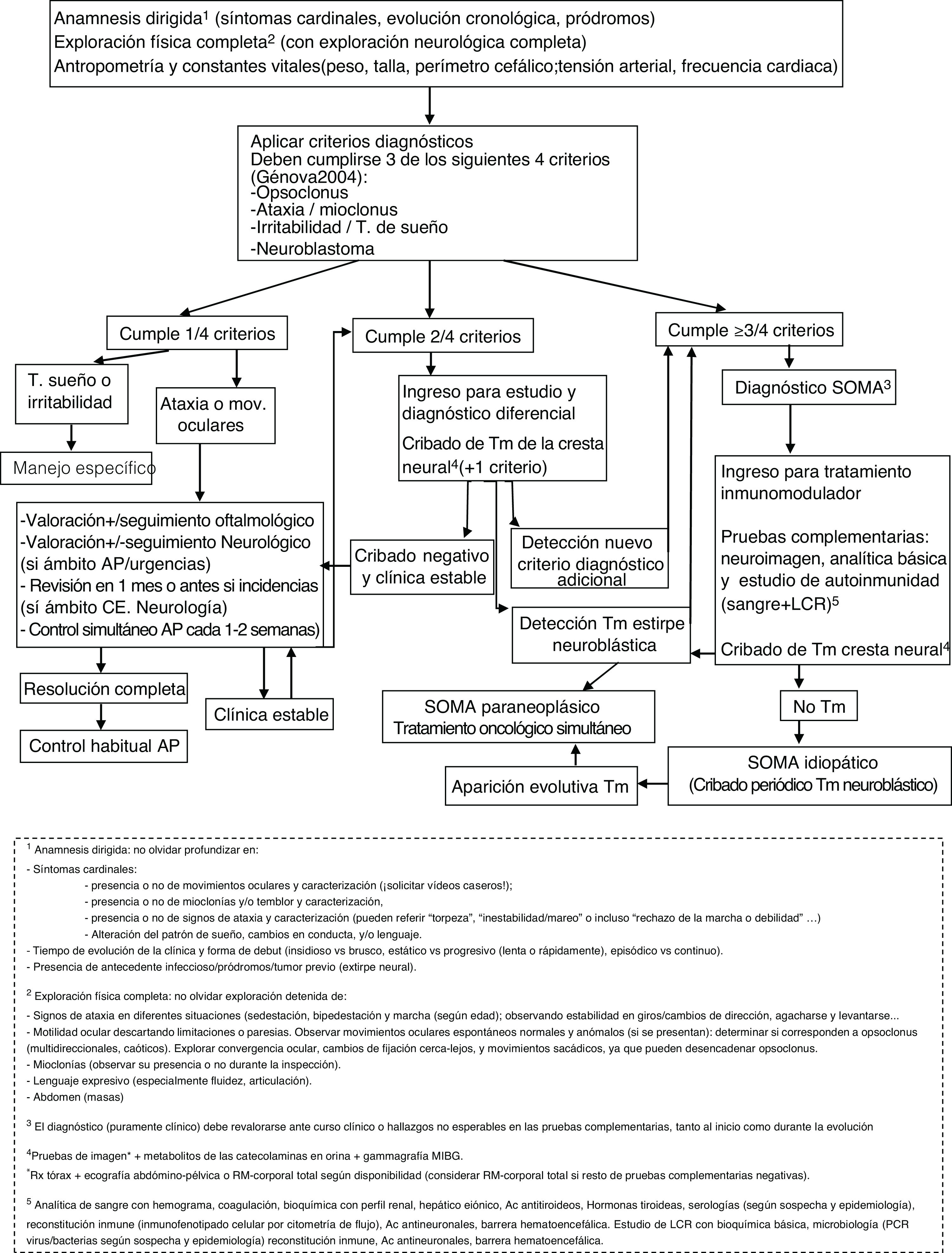
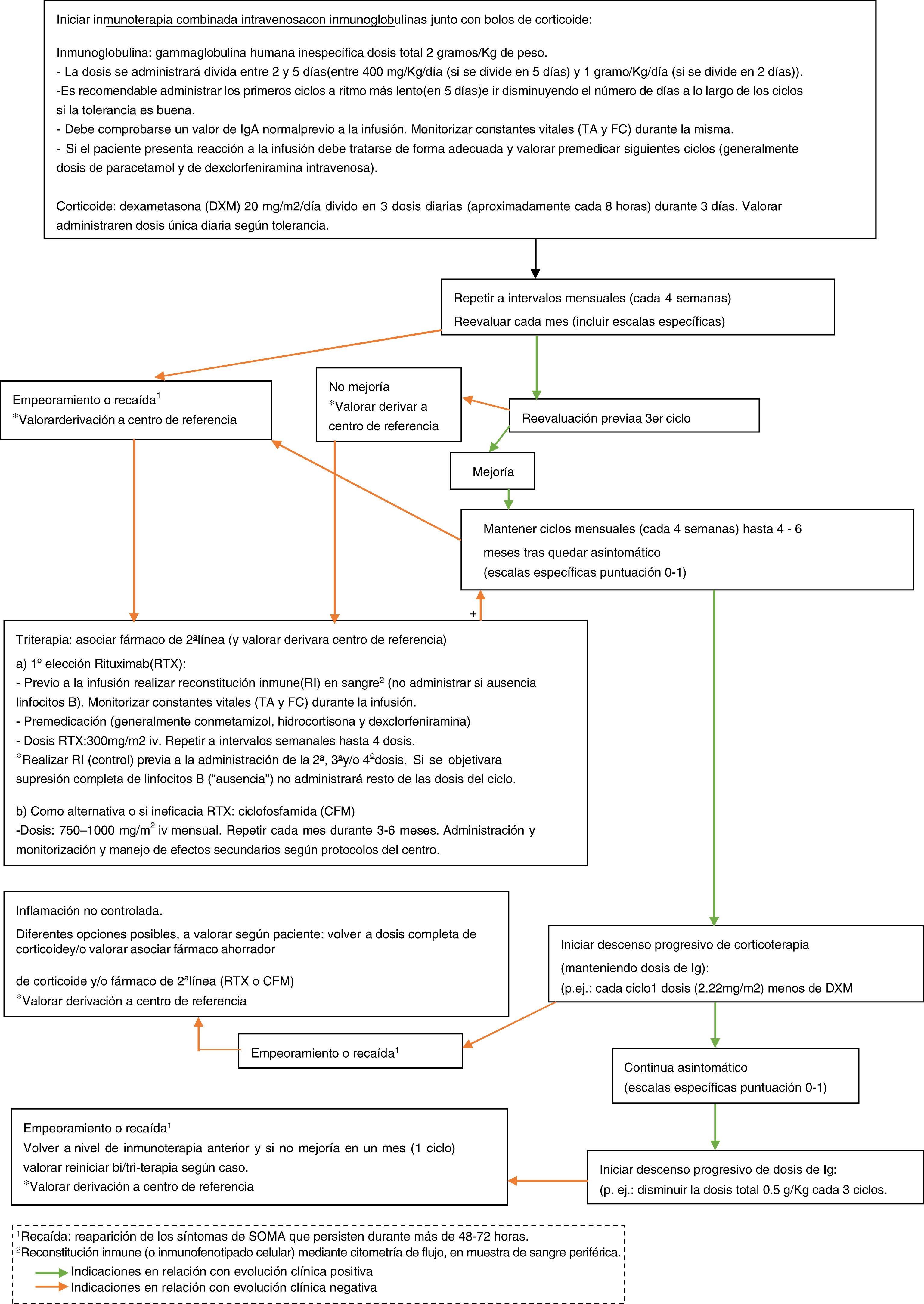
ConclusionDespite the small sample size, retrospective approach, and lack of a standard management protocol, our findings seem to show a certain association between treatment approaches and neurocognitive progression. The principal requirement for early treatment is early diagnosis, for which recognising symptoms is crucial. We would also highlight the importance of actively searching for other suggestive signs and symptoms (which may be exclusively behavioural) in patients presenting ataxia, which should lead to a high level of suspicion. Once more, we observed a relevant association with neuroblastic tumours, which generally present limited aggressiveness; in the diagnosis of these tumours, it is essential to use such imaging techniques as CT or MRI, given the high rate of false negatives in other tests that rely on metabolic activity or tumour size (Fig. 1 shows a diagnostic algorithm for suspected OMAS). The presence or absence of sequelae may have been influenced by the intensification of immunotherapy in cases of incomplete clinical response, as well as the early onset of treatment. Therefore, we stress the importance of administering combination therapy from onset, with tritherapy in cases of insufficient treatment response (Fig. 2 shows the therapeutic algorithm for OMAS). In this regard, the development and implementation in paediatric neuroimmunology departments of such treatments as rituximab, which are more targeted against the suspected underlying inflammatory mechanism, has in our opinion been a highly positive change. The study and implementation of new immune markers and targeted treatments, and the development of management protocols, may help in efforts to achieve optimal management and prognosis in OMAS.

Diagnostic algorithm for suspected opsoclonus-myoclonus-ataxia syndrome. Ab: antibodies; CSF: cerebrospinal fluid; EC: external consultations; IR: immune reconstitution; MIBG: metaiodobenzylguanidine; MRI: magnetic resonance imaging; RX: radiography; OMAS: opsoclonus-myoclonus-ataxia syndrome; PC: primary care; PCR: polymerase chain reaction.

This study has received no specific funding from any public, private, or non-profit organisation.
Conflicts of interestThe authors have no conflicts of interest to declare.
