




El síndrome opsoclono-mioclono-ataxia es un raro trastorno de inicio pediátrico; de base neuroinflamatoria y origen paraneoplásico, parainfeccioso o idiopático. Actualmente no hay biomarcadores, siendo el diagnóstico clínico. El pronóstico cognitivo parece estar relacionado con el inicio temprano de la terapia inmunomoduladora.
MétodoSe describen las características epidemiológicas, clínicas, terapéuticas y pronósticas a largo plazo de una cohorte de 20 pacientes españoles.
ResultadosLa edad media de debut fue de 21 meses (2-59 meses). La ataxia y el opsoclonus fueron los síntomas de inicio más frecuentes y predominantes en la evolución. El tiempo medio desde los primeros síntomas hasta el diagnóstico fue de 1,1 mes. Un tumor de extirpe neuroblástica fue detectado en el 45%, realizándose resección quirúrgica en siete y quimioterapia en dos pacientes. En el estudio de líquido cefalorraquídeo se constató pleocitosis en cuatro (25%), con negatividad de anticuerpos antineuronales y bandas oligoclonales en todos los casos estudiados. En el 100% se emplearon fármacos inmunomoduladores. En nueve pacientes el tratamiento combinado inmunomodulador se inició desde el momento del diagnóstico, y en cinco el tiempo medio de implementación fue de 2,2 meses. A largo plazo, seis de 10 pacientes con seguimiento superior a cinco años presentaban secuelas cognitivas leves o moderadas; cuatro pacientes presentaron recaídas, generalmente coincidiendo con el descenso de la corticoterapia.
ConclusionesEl inicio precoz de la inmunoterapia, así como de la triple terapia en los casos que lo precisaron, se relacionó con una menor frecuencia de afectación cognitiva a los dos años del debut.
Opsoclonus-myoclonus-ataxia syndrome is a rare neuroinflammatory disorder with onset during childhood; aetiology may be paraneoplastic, para-infectious, or idiopathic. No biomarkers have yet been identified, and diagnosis is clinical. Better cognitive prognosis appears to be related to early onset of immunomodulatory therapy.
MethodsWe describe the epidemiological, clinical, therapeutic, and long-term prognostic characteristics of a cohort of 20 Spanish patients.
ResultsThe mean age of onset was 21 months (range, 2-59). Ataxia and opsoclonus were the most frequent symptoms both at disease onset and throughout disease progression. The mean time from onset to diagnosis was 1.1 months. Neuroblast lineage tumours were detected in 45% of patients; these were treated with surgical resection in 7 cases and chemotherapy in 2. Cerebrospinal fluid analysis revealed pleocytosis in 4 cases (25%) and neither antineuronal antibodies nor oligoclonal bands were detected in any patient. Immunomodulatory drugs were used in all cases. Nine patients started combined immunomodulatory treatment at the time of diagnosis, and 5 patients after a mean of 2.2 months. In the long term, 6 of the 10 patients followed up for more than 5 years presented mild or moderate cognitive sequelae. Four patients presented relapses, generally coinciding with the decrease of corticosteroid doses.
ConclusionsEarly initiation of immunotherapy, as well as triple combination therapy, where needed, was associated with a lower frequency of cognitive impairment 2 years after onset.
El síndrome opsoclono-mioclono-ataxia (SOMA) es una enfermedad de probable base neuroinmunológica, definida clínicamente por la presencia de síntomas motores, considerándose cardinales la ataxia, el mioclono (sacudidas musculares de origen central) y el opsoclono (movimientos oculares erráticos y multidireccionales). Esta triada aparece generalmente acompañada de irritabilidad y/o insomnio en diferentes combinaciones1. Esta clínica puede estar asociada o no a la existencia de un tumor de la cresta neural (fundamentalmente neuroblastoma) constituyendo otro de los elementos definitorios de la enfermedad1,2. Algunos autores han tratado de sintetizarlo para simplificar su diagnóstico en la presencia de tres de los siguientes síntomas o signos: opsoclono o fluter ocular, mioclono y/o ataxia, neuroblastoma, cambios de humor y/o trastorno del sueño con frecuencia asociado a marcada irritabilidad1,2. Se ha estimado una incidencia de 0,18 casos por millón de habitantes3 o 0,27-0,40 casos por millón de niños4.
El SOMA se asocia con tumores de la cresta neural hasta en el 50% de los casos, entre ellos neuroblastoma, ganglioneuroblastoma y ganglioneuroma. Algunos autores especulan que este porcentaje puede ser mucho mayor, pero una respuesta inmunológica, humoral o celular, provocaría la destrucción de la masa tumoral antes de que pudiese ser detectada5. En ausencia de tumor, se ha establecido una relación temporal con diversas infecciones, entre ellas el virus de Epstein-Barr6, Mycoplasma pneumoniae7,8, virus West Nile9, hepatitis C10, adenovirus C311 o rotavirus12. No obstante, en muchos casos es difícil establecer una verdadera causalidad, por lo que la etiología se consideraría «idiopática». En todos los casos de SOMA, una base inmunomediada parece la responsable de la sintomatología cardinal por afectación a nivel no sólo de tronco encéfalo o cerebelo, sino también supratentorial13.
Se ha publicado un elevado porcentaje de pacientes con secuelas neurocognitivas y psiquiátricas, aunque, en los últimos años, el pronóstico ha mejorado con el uso temprano de terapias combinadas14. A pesar de esto, existen aún controversias sobre la influencia del retraso diagnóstico o terapéutico en sí mismo sobre estos aspectos cognitivos-conductuales15–17.
Se presenta una serie pediátrica española de 20 pacientes con SOMA mostrando sus características epidemiológicas, clínicas, etiológicas, terapéuticas y de evolución a largo plazo.
Material y métodosPacientesSe obtuvieron datos de los pacientes menores de 18 años diagnosticados de SOMA en hospitales de seis ciudades españolas (Madrid, Pamplona, Valladolid, San Sebastián, Vitoria y Granada) desde 2006 hasta 2018. La información fue recogida mediante un cuestionario estandarizado.
Se tomó como criterio de inclusión el diagnóstico de SOMA cuando cumplía tres de los cuatros criterios: 1) opsoclono o fluter ocular, 2) neuroblastoma, 3) mioclono y/o ataxia, y 4) cambios de humor y/o trastorno del sueño, con frecuencia asociado a marcada irritabilidad.
Datos clínicosSe analizaron datos correspondientes al sexo, edad de inicio, signos y síntomas propios de SOMA, edad al diagnóstico, cronología y evolución de los síntomas, tiempo de retraso diagnóstico y de inicio de tratamiento, exploración física con escalas específicas (Pranzatelli y Mitchel)18,19, pruebas complementarias (despistaje infeccioso, inmunológico y tumoral), tratamientos recibidos y forma de implementación, así como evolución a largo plazo desde un punto de vista cognitivo-conductual, motor y de trastorno del sueño.
Análisis estadísticoLa homogeneidad de las variables demográficas y los parámetros clínicos señalados fueron analizados durante el estudio. La descripción se realizó mediante la media, mediana, rango y desviación típica de las variables cuantitativas, así como con la frecuencia absoluta y frecuencia relativa de las variables cualitativas. Para las variables cuantitativas se emplearon pruebas no paramétricas (Rho de Spearman) dado el tamaño muestral. Las variables cualitativas fueron analizadas mediante test de homogeneidad basadas en la distribución χ2 cuando los valores esperados lo hicieron posible y mediante test exactos de Fisher, en caso contrario. Se consideró significación estadística valores de p < 0,05. Se analizaron mediante el paquete estadístico SPSS versión 17.0 IBM inc.
ResultadosCaracterísticas clínicas y epidemiológicasSe analizaron un total de 20 pacientes con diagnóstico definitivo de SOMA, cuyas características epidemiológicas y forma de presentación clínica en el momento del debut se encuentran resumidas en la tabla 1. El tiempo de seguimiento ha sido desde tres meses (última paciente diagnosticada) hasta 12 años, con una mediana de siete años.
Características al debut en 20 pacientes con SOMA
| Características clínicas | N | % | Media (DE) | Rango | Descripción/comentarios |
|---|---|---|---|---|---|
| Sexo femenino | 11 | 55 | |||
| Edad media al diagnóstico (m) | 21 (11,8) | 2-59 m-90% con 14-30 m-10% con ≤ 6 m | Similar en niñas (21 m) y niños (21,3 m) | ||
| Retraso diagnóstico (m) | 1,1 (1,78) | ||||
| Síntomas prodrómicosTotalCon agente infeccioso | 10 | 50 | 5 infección respiratoria (IRVA / OMA / bronquitis), 2 GEA, 2 fiebre sin foco, 1 exantema urticariforme. | ||
| 3/10 | 33 | Mycoplasma pneumoniae IgM, Rinovirus (nasof), VRS (nasof) | |||
| Primer síntoma en aparecerAtaxiaOpsoclono | 146 | 7030 | |||
| Sint. cardinales evolutivosAtaxiaIrritabilidadOpsoclonoT. sueñoMioclonoT. habla | 19181716136 | 959085806530 | 7/18 (39%) precisaron farmacoterapia10/16 (62,5%) precisaron farmacoterapia (melatonina) |
% (porcentaje sobre el total de casos de la muestra); DE (desviación estándar); GEA (gastroenteritis aguda); IgM (inmunoglobulina M); IRVA (infección respiratoria de vías altas); m (meses); N (número absoluto); nasof (nasofaríngeo); OMA (otitis media aguda); sint (síntomas); T. (trastorno).
La sintomatología cardinal del SOMA (opsoclono, mioclono, ataxia, irritabilidad e insomnio) no estaba presente en todos los casos. Los primeros síntomas en aparecer fueron la ataxia y el opsoclono, que fueron también los más frecuentes en la evolución junto con la irritabilidad. De los pacientes mayores de dos años (n = 6), un tercio no presentaron opsoclono, mientras que sólo estaba ausente en uno de los 14 pacientes con inicio anterior a esta edad (7%). En tres pacientes, en los que no se refería un mioclono claro, se indicaba temblor. En el momento del ingreso, sólo cinco pacientes (25%) cumplían los criterios clínicos diagnósticos. Según la duración del SOMA desde el inicio hasta la resolución de todos los síntomas, fue agudo (≤ tres meses) en cuatro pacientes, subagudo (tres a 12 meses) en cinco pacientes y crónico (≥12 meses) en ocho pacientes. En tres pacientes no se conoce con exactitud el tiempo de cese de la clínica.
En el momento inicial, sólo seis de los pacientes (30%) tenían realizada una exploración sistemática con la escala de Mitchel y en 10 (50%) con la de Pranzatelli, llevando un seguimiento con las mismas a más largo plazo (seis, 12 y 24 meses) en seis (30%) y siete (35%) casos, respectivamente. En el momento del debut, la puntuación media en la escala de Mitchell fue de 11,5 (DE 1,1) y en la de Pranzatelli de 21,8 (DE 2,52). Teniendo en cuenta el valor de esta última (en los que se realizó), sólo un caso presentaba una afectación leve (considerándose puntuación de 0 a 12), cinco moderada (puntuaciones de 13 a 24) y cuatro grave (de 25 a 36).
Ocho pacientes (40%) tenían una evaluación neuropsicológica en el momento del diagnóstico. En los menores de dos años incluía la escala de desarrollo de Batelle y en los mayores las escalas de inteligencia de Weschler, en su versión para preescolar y primaria (WPPSI) o bien para niños (WISC) según la edad. Además, en algunos pacientes se habían aplicado adicionalmente otras pruebas de forma puntual a lo largo de su evolución (entre ellas la escala de McCarthy de aptitudes y psicomotricidad (MSCA), la batería de evaluación para niños de Kaufman (K-ABC), el cuestionario de madurez neuropsicológica infantil (CUMANIN) o el test de ejecución continua de Conners (CPT) en aquellos con dificultades atencionales). En la valoración neuropsicológica al diagnóstico se evidenciaron alteraciones en cinco de ellos (62%): tres en forma de retraso global del desarrollo leve (cocientes de desarrollo (CD) en la escala de Batelle entre 76 y 86), uno junto con alteración de conducta y atención; otro paciente con alteraciones de atención y motricidad, y el último caso mostró retraso del lenguaje y aprendizaje global. En el seguimiento, y excluyendo al último paciente con un tiempo de evolución menor de un año, sólo seis pacientes (30%) tenían nuevas evaluaciones cognitivo-conductuales.
Caracterización de la enfermedad oncológicaEn el 45% de los pacientes (n = 9) se puso de manifiesto la existencia de un tumor de origen neuroblástico (ver características y manejo terapéutico en tablas 2 y 3). En dos casos, el tumor se detectó antes del diagnóstico del SOMA, desarrollándose este cuadro entre los ocho y 24 meses posteriores, sin que se objetivase recaída tumoral. La edad media de aparición del neuroblastoma fue algo inferior que en el caso del ganglioneuroblastoma. No se encontró un retraso diagnóstico de la etiología tumoral, hallándose durante el primer despistaje, que incluyó el estudio de metabolitos de las catecolaminas en orina, pruebas de imagen y gammagrafía con metayodobencilguanidina (MIBG). Entre las pruebas de imagen se realizaron, en diferentes combinaciones: radiografía (Rx) de tórax, ecografía abdominal; así como tomografía computarizada (TC) y/o resonancia magnética (RM) de forma dirigida (sí se detectó previamente una masa sospechosa en otra prueba) y/o corporal total. Estas últimas detectaron el tumor en todos los casos, incluso aquellos en que no se había detectado inicialmente mediante Rx tórax y ecografía y aquellos con MIBG y estudio de catecolaminas negativas.
Caracterización de la enfermedad oncológica en 9 pacientes con SOMA paraneoplásico
| Características de la enfermedad oncológica | N | % | Descripción/comentarios | |
|---|---|---|---|---|
| Tipo de tumor | Neuroblastoma (NB) | 7 | 78 | Edad media al diagnóstico (NB) 14,7 m (rango de 5 a 27 m) |
| Ganglioneuroblastoma | 2 | 22 | Edad al diagnóstico 20 y 30 m respectivamente | |
| Localización | Adrenal o abdominal/paraespinal | 6 | 67 | |
| Mediastino posterior | 1 | 11 | ||
| Paravertebrales (sin especificar) | 2 | 22 | ||
| Pruebas complementarias | Catecolaminas orina | 9 | 100 | Alterado en 3/9 (33%) |
| Gammagrafía MIBG | 8 | 89 | Captación patológica en la zona tumoral en 3/8 (37,5%) | |
| RM/TC dirigido/corporal total | 9 | 100 | Objetiva masa tumoral en 9/9 (100%) incluso en casos con Rx tórax y ecografía abdominal previas normales +/- Catecolaminas orina y Gammagrafía MIBG negativas | |
| Tratamiento | Cirugía resectiva completa | 7 | 78 | 2 de ellos con debut de SOMA 8 y 24 meses después sin evidenciar recidiva |
| Quimioterapia | 2 | 22 | Uno de ellos aislada por no operable, otro tras cirugía |
% (porcentaje sobre el total de casos paraneoplásicos); MIBG (metayodobencilguanidina); N (número absoluto); RM (resonancia magnética); Rx (radiografía); TC (tomografía computarizada).
Características comparativas al debut y en el curso evolutivo de los diferentes grupos (paraneoplásicos e idiopáticos/criptogénicos asociados o no a infección)
| Variables de interés | Paraneoplásicos | Idiopáticos/criptogénicos | |
|---|---|---|---|
| Presumiblemente parainfecciosos | Sin antecedente infeccioso conocido | ||
| Número de pacientes | 9 | 7 | 4 |
| Edad media de debut | 20,22 | 26,42 | 14 |
| Pródromos | 3 (33%) | 7 (100%) | 0 |
| Tiempo hasta cumplir criterios diagnósticos (3 de 4) (meses) | 3,12 | 0,63 | 1,9 |
| Necesidad de fármacos 2 ª línea | 3 (33%) | 1 (14%) | 3 (75%) |
| Asintomáticos actualmente | 6 (66%) | 5 (55,5%) | 1 (25%) |
| Persiste clínica actualmente | 2 (22,2%) | 0 | 3 (75%) |
| Pérdida de seguimiento | 1 | 2 | 0 |
| Tiempo medio para la resolución completa (meses) a | 19,1 | 3,4 | 30 |
| Afectación cognitiva y/o conductual a 2 años b | 4 | 1/6(b 1) | 2/2(b 2) |
| Trastorno de sueño a 2 años | 1 | 0/6(b 1) | 1/2(b 2) |
| Otras secuelas a los 2 años | Ataxia residual (1) | 0/6 | Torpeza motora leve (2/4) |
Siete de los casos en los que se documentó una infección clara en las semanas previas al inicio del cuadro clínico de SOMA (cinco infecciones respiratorias y dos gastrointestinales), y no habiendo detectado otra etiología relacionada ni en el debut ni posteriormente, se han considerado presumiblemente parainfecciosos (ver características en tabla 3). Destacamos la mayor rapidez de resolución de la clínica en este grupo con menor necesidad de tratamiento de segunda línea.
NeuroinflamaciónEn 16 pacientes (80%) se llevó a cabo una punción lumbar, evidenciando pleocitosis (leucocitos ≥ 4/mm3) en cuatro (25%). La glucorraquia fue normal en todos los pacientes, uno presentó hiperproteinorraquia de 85 mcg/L (valores normales 15-45). Se realizó estudio de barrera hematoencefálica con determinación de bandas oligoclonales en ocho casos (50%), y sólo un paciente, presumiblemente parainfeccioso por Mycoplasma pneumoniae, tuvo una banda en el espectro de las beta-globulinas. En nueve de 17 casos (53%), se estudió la presencia de diferentes anticuerpos antineuronales en suero, LCR o en ambos, no evidenciando ningún resultado positivo.
Tratamiento inmunomoduladorTodos los pacientes recibieron algún tipo de tratamiento inmunomodulador. En la mayoría de los casos se instauró en el momento del diagnóstico, siendo el tiempo medio desde el inicio de los síntomas hasta la instauración de la inmunoterapia asimilable al tiempo de retraso diagnóstico (1,1 mes; DS 1,78 meses). En cuanto al tiempo que pudo transcurrir entre el diagnóstico definitivo y el inicio del tratamiento, en los que se documentó, el tiempo medio fue de 0,3 meses (0 días-1,5 meses). En cuanto a la modalidad de tratamiento, la monoterapia incluyó corticoides o ACTH (ningún paciente recibió exclusivamente inmunoglobulinas). Se realizó un tratamiento combinado en 14 casos (70%). Ocho pacientes recibieron fármacos de segunda línea (seis rituximab, dos ciclofosfamida), en siete añadido a una biterapia previa de inmunoglobulina y corticoides y en uno añadido a corticoides.
En nueve niños el tratamiento combinado inmunomodulador se inició desde el momento del diagnóstico, y en los cinco restantes, el tiempo medio de implementación fue de 2,2 meses (entre 15 días y cuatro meses). En seis pacientes fue necesario el uso de ≥ 3 terapias en combinación para el control clínico. El tiempo transcurrido desde el inicio de una monoterapia o biterapia hasta la introducción de un fármaco de segunda línea, fue de ocho meses (de tres a 26 meses).
La media de ciclos de inmunoglobulinas fue de 14 (rango 1 a 36). Seis pacientes utilizaron rituximab y en dos se necesitó repetir un nuevo ciclo de cuatro dosis a 375 mg/m2. Los dos pacientes en los que se empleó ciclofosfamida recibieron dosis mensuales (tres y ocho dosis, respectivamente). En cuanto al tiempo de mantenimiento del tratamiento inmunomodulador (globalmente): no se pudo precisar en cuatro de ellos; cuatro mantienen inmunoterapia actualmente (entre tres y 27 meses). Los 12 restantes la mantuvieron un tiempo medio de 17.6 meses (entre uno y 36 meses).
Curso evolutivo, recaídas y secuelasDe los ocho casos con evolución crónica (≥ 12 meses desde el debut de SOMA), cuatro quedaron asintomáticos entre los 17 y 55 meses de seguimiento sin recidivas posteriores, en los cuatro restantes persiste clínica llevando en la actualidad entre 13 meses y 11 años de evolución. Estos cuatro mantienen ataxia; dos pacientes (tiempo de evolución menor, de 12 y 24 meses) la presentan de forma mantenida, en uno de ellos, con irritabilidad. Los otros dos (con tiempo de evolución en torno a 10 años) presentan leve ataxia intermitente (en contexto febril o infeccioso). De forma global, los 12 casos asintomáticos, alcanzaron este estado en un tiempo medio de 11,72 meses (rango 0,5 a 55 meses), siendo variable el tiempo hasta la resolución de los síntomas individualmente (tabla 4).
Curso evolutivo de los síntomas cardinales
| Síntoma | Casos con resolución completa (N/n total; %) | Tiempo hasta la resolución completa (media ± DE; rango) | Comentarios |
|---|---|---|---|
| Opsoclono | 17/17 (100%) | 3,7 ± 3,4 meses (0,05-10 meses) | |
| Mioclono | 13/13 (100%) | 4,7 ± 3,7 meses | |
| Ataxia | 15/19 (78,9%) | 9 ± 7,9 meses, mediana de 5 meses | Síntoma que más tardó en resolverse |
| Irritabilidad | 15/18 (83%) | Mediana 5 meses | Un caso con trasformación evolutiva a T. de conducta |
% (porcentaje de pacientes en que se resolvió el síntoma sobre el total que lo presentó); DE (desviación estándar); n (número de pacientes de la muestra que presentó el síntoma); N (número absoluto de pacientes de la muestra en que se resolvió completamente el síntoma); T (trastorno).
A pesar de ser las secuelas cognitivas y conductuales las más frecuentemente observadas de manera clínica (tabla 5), sólo pudo determinarse la afectación inicial y su evolución de una manera objetiva en los seis pacientes que disponían de evaluación neuropsicológica al debut y controles seriados posteriores. En dos de estos seis, que presentaban una afectación inicial global leve (CD 73 y 86 en la escala de desarrollo de Batelle), se constató mejoría hasta la normalización (seguimiento a los dos años del debut, con cociente intelectual total de 94 y 106, respectivamente en la escala de inteligencia de Wechsler). Los dos que presentaban alteración atencional en la primera evaluación, mantuvieron alteraciones en las funciones ejecutivas en los controles (trastorno por déficit de atención e hiperactividad [TDAH]) (seguimiento a cinco y 10 años, respectivamente). Uno de ellos dispone de pruebas atencionales de control tras iniciar tratamiento con estimulantes objetivándose normalización de los parámetros específicos. Dos pacientes desarrollaron evolutivamente un trastorno específico del aprendizaje de la lectoescritura, uno con evaluación neuropsicológica inicial normal y el otro sobre un retraso del lenguaje, pero con evolución favorable a los 10 años de seguimiento.
Secuelas
| Tiempo de seguimiento | N | N sec | Diagnóstico (secuela) |
|---|---|---|---|
| > 10 años | 3 | 3 | Dificultades en la lectoescrituraDificultades en la lectoescritura + TDAHTDAH con predominio de la hiperactividad |
| 5-10 años | 7 | 4 | TDAH (2 casos)Dificultades en la lectoescrituraTorpeza global con ataxia leve residual |
| 2-5 años | 7 | 2 | Irritabilidad persistenteRetraso del lenguaje |
| < 2 años | 3 | No valorable por poco tiempo de evolución, y 2 de ellos manteniendo clínica cardinal aguda |
N (número absoluto de pacientes de la muestra con el tiempo de seguimiento indicado); N sec (número de pacientes con secuelas con el tiempo de seguimiento indicado); TDAH (trastorno por déficit de atención e hiperactividad).
Cuatro de los pacientes (25%) presentaron recaídas claras. Tres de ellos eran pacientes con neuroblastoma intervenido al debut con resección completa y con pruebas de despistaje tumoral repetidamente negativas en la evolución (tiempo de seguimiento de 7,5, 9 y 9,5 años, respectivamente). De los cuatro, dos habían recibido monoterapia y los otros dos, tratamiento combinado. Uno de ellos, que había recibido fármacos de segunda línea recayó tras dos años asintomático. Se reinició inmunoterapia con inmunoglobulinas junto con un segundo ciclo de rituximab, quedando asintomático pocos meses después, sin presentar nuevas recidivas. El otro presentó dos recidivas, ambas tras suspensión de corticoides (prednisona) que recibía en combinación con inmunoglobulinas; precisando un ciclo de rituximab y asociar azatioprina. Respecto a los casos que recibieron monoterapia, en uno de ellos reapareció la clínica al iniciar la retirada de ACTH (monoterapia) a los cuatro meses del debut, tras una respuesta completa inicial a dicho tratamiento. El otro (único idiopático) recidivó cinco meses tras la suspensión de corticoides (prednisona) reiniciándose a dosis completas con resolución clínica.
Estudio de posibles condicionantes pronósticosEn cuanto a las posibles implicaciones pronósticas de la precocidad y gravedad al debut, en nuestra serie se encontró una relación estadísticamente significativa (Rho de Spearman 1, p = 0,01) entre la mayor gravedad clínica inicial según la escala de Mitchell y el tiempo hasta la resolución clínica completa. Sin embargo, no ocurrió lo mismo al aplicar la escala de Pranzatelli. Debe tenerse en cuenta que sólo en seis pacientes se disponía de la puntuación inicial en la escala de Mitchell y en 10 la de Pranzatelli (en tres se reflejaban ambas), lo que pudo influir en los resultados. Por otro lado, se halló cierta relación entre una menor edad al debut y un mayor tiempo de duración de la clínica, sin ser esta estadísticamente significativa.
Se consideró debut precoz aquel anterior a los 18 meses, por corresponder a un momento del desarrollo neurológico en que la mayoría de los niños normales ya han adquirido la marcha autónoma e iniciado el lenguaje expresivo verbal. No hubo diferencias significativas en la frecuencia de afectación cognitiva a los dos años del debut entre aquellos de debut precoz y los de inicio posterior (16% vs. 36%, p = 0,6). Sólo dos pacientes tenían un seguimiento inferior a dos años, los cuales no se incluyeron en este análisis. No se analizó la asociación con afectación cognitiva a los cinco años dado que sólo 10 pacientes disponen de un seguimiento suficiente. El grupo de inicio clínico precoz precisó asociación de fármacos de segunda línea con mayor frecuencia (62,5% vs. 25%), sin ser la diferencia estadísticamente significativa (p = 0,16).
En cuanto a la importancia de la precocidad del tratamiento, en nuestra serie no hubo una relación estadísticamente significativa entre el tiempo desde el debut clínico hasta la instauración de inmunoterapia y la puntuación en las escalas clínicas (de Mitchell y de Pranzatelli) a los seis, 12 y 24 meses. Si bien, solamente 11 disponían de puntuaciones seriadas de alguna de las dos (sólo seis ambas), lo que pudo influir en el análisis. Tampoco hubo correlación entre este retraso terapéutico y el tiempo hasta quedar asintomático (correlación de Pearson 0,25, p = 0,42). La instauración de un tratamiento inmunomodulador precoz (antes de dos meses tras el debut clínico) en cualquier modalidad se asoció con una menor frecuencia de afectación cognitiva a los dos años (13% vs. 50%) sin ser la diferencia estadísticamente significativa, pero sí clínicamente relevante.
En los pacientes que precisaron la asociación de un fármaco de segunda línea, el retraso en su inició se relacionó con el retraso en la resolución de la clínica de SOMA (Rho de Spearman 0,95, p = 0,05). No hubo diferencias significativas en la frecuencia de afectación cognitiva entre los que recibieron fármacos de segunda línea y los que no (33% vs. 27%). Sin embargo, sí que hubo una relación con la precocidad de la intensificación. De los ocho pacientes que recibieron un fármaco de segunda línea, cuatro recibieron la primera dosis en los primeros seis meses de tratamiento inmunoterápico (dos a los tres meses) y tres después del sexto mes (dos a los 10 meses y uno a los 26). Seis pacientes recibían previamente tratamiento combinado en biterapia (esteroides e inmunoglobulinas), por lo que la adición del fármaco de segunda línea supuso el inicio de la triterapia. Todos los pacientes (3/3) que iniciaron triterapia en los primeros seis meses de tratamiento mantuvieron un estado cognitivo normal a los dos años del debut, mientras que todos (3/3) los que lo hicieron después del sexto mes de tratamiento presentaban afectación cognitiva en ese tiempo de evolución.
DiscusiónLa presente es la cohorte pediátrica española con mayor número de casos de SOMA con seguimiento a medio-largo plazo que hemos encontrado publicada hasta el momento. Esto nos permite hacer algunas observaciones epidemiológicas, clínicas y evolutivas sobre el manejo de esta infrecuente enfermedad en nuestro medio, que pueden ser útiles en la práctica clínica presente y futura. Debemos tener en cuenta que, dada la baja frecuencia de esta entidad, el número de pacientes de nuestra serie sigue siendo limitado para establecer conclusiones sólidas en algunos aspectos.
Características clínicas y epidemiológicasEn nuestra cohorte se objetivó un ligero predominio de SOMA en el sexo femenino (55% del total), muy similar al hallado en otras cohortes donde se dan porcentajes entre 52 y 65%1,4,20,21. El factor de riesgo que parece suponer el sexo femenino, al igual que en otras entidades autoinmunes/autoinflamatorias continúa siendo objeto de estudio. La edad de presentación predominante fue la preescolar, entre 14 y 30 meses con dos pacientes (10%) < seis meses.
El tiempo medio de retraso diagnóstico (1,1 mes), fue muy similar a los últimos datos de Pranzatelli et al.1, que lo sitúan en 1,2 meses y mejor que los dados por Singhi et al.22, con dos meses (entre dos días y 12 meses) o Huddar et al.23 que hablan de 3,8 ± cinco meses (entre 15 días y 16 meses). Esto puede ser clave a la hora de una instauración precoz de tratamiento, despistaje de patología tumoral, incluso evolución a largo plazo15,16.
El inicio con ataxia aislada (síntoma de debut más frecuente)1,24 dificulta y retrasa el diagnóstico al plantearnos el diagnóstico diferencial principalmente con la ataxia aguda postinfecciosa22. Si bien esta última entidad afecta menos a lactantes, con mayor frecuencia de pródromos y aparición de exantema, generalmente por su asociación con el virus de la varicela zóster, principalmente24,25. La aparición de otros síntomas conductuales como irritabilidad y/o insomnio llamativo junto a una ataxia persistente debe plantear la posibilidad de un SOMA, habiendo ocurrido esto en un tercio (2/6) de los mayores de dos años en nuestro estudio, ambos asociados a neuroblastoma. En algunos de los pacientes sin mioclono se documentó temblor. Según descripciones previas, el mioclono puede adquirir en casos leves una apariencia trémula que podría confundirse con el temblor, aunque en general las sacudidas son más intensas y se induce más claramente por la acción1.
En la evaluación y seguimiento clínico de los pacientes sería deseable el empleo de escalas específicas que permitiesen una estratificación del grado de afectación, una valoración más objetiva de la respuesta al tratamiento, la aparición de recidivas y establecer comparaciones y posibles relaciones entre gravedad clínica y variables relacionadas con el pronóstico. En este sentido, debemos destacar negativamente en nuestra serie la baja proporción de pacientes evaluados sistemáticamente mediante dichas escalas. De igual modo, a pesar de la frecuencia de la afectación cognitiva y sus potenciales repercusiones, sólo un 30% (6/20) de la muestra tenía valoración neuropsicológica. Esto puede estar relacionado con la dificultad de acceso de muchos hospitales españoles a equipos de Neuropsicología Infantil.
Caracterización de la enfermedad oncológicaLa frecuencia, estirpe y predominio etario (a edad algo mayor el ganglioneuroblastoma) de los casos paraneoplásicos fue similar a lo reportado en series mundiales previas1. De acuerdo con lo descrito anteriormente26–29 el pronóstico de la enfermedad oncológica fue favorable, con curación completa en todos los casos (9/9). Cuando hubo tumor, este se detectó en el primer despistaje en todos los casos. Si bien, dado que algunos autores especulan que el tumor podría retroceder espontáneamente o ser lo suficientemente pequeño como para evadir la detección, la designación del resto de los casos «no paratumorales», en cierto modo, sería incierta20,28.
En contraste con la alta sensibilidad descrita previamente para la MIBG en el diagnóstico de neuroblastoma (en torno a 95%)30, en nuestra serie fue menor en comparación con la TC o la RM, coincidiendo con lo reportado en otros estudios22,24,31,32. Esta menor sensibilidad de la MIBG se ha atribuido al bajo grado de malignidad/agresividad del tumor en cuestión22,23,31,32. Asimismo, Biasotti et al.30 encuentran una asociación entre MIBG negativa y normalidad de catecolaminas en orina, que relaciona con la existencia de neuroblastomas no secretores (en torno al 5%). Dado que los tumores asociados a la aparición de SOMA son frecuentemente de bajo grado, el diagnóstico basado en pruebas que dependen de la actividad metabólica (como la MIBG y el perfil de metabolitos de catecolaminas en orina) puede no ser lo suficientemente sensible si se emplean de forma aislada; deberían por tanto acompañarse de pruebas de imagen protocolizadas31. Asimismo, por su menor tamaño pueden no ser detectados por imágenes de radiografía simple o ecografía, siendo necesarias técnicas más meticulosas con múltiples cortes como la TC o la RM16. Por todo lo anterior, el uso de TC corporal o mejor, RM corporal total (por su menor radiación) creemos que debería plantearse como la herramienta diagnóstica con mayor coste-efectividad en estos pacientes. Destacamos que algunos de los pacientes paraneoplásicos presentaron síntomas infecciosos previos, por lo que esto no debe descartar la etiología paratumoral6,24.
Caracterización de los casos «idiopáticos/criptogénicos»La etiología «presumiblemente infecciosa» se asoció, en nuestra serie, a un mejor pronóstico en cuanto a una resolución clínica más rápida, precisando fármacos de segunda línea con menos frecuencia y con menor porcentaje de secuelas a los dos años (tabla 3)6,24.
Caracterización a nivel inmunológicoSólo en el 50% de los casos se realizaron bandas oligoclonales, como posible expresión del papel patogénico de los linfocitos B33, hallando una banda en un solo caso con neuroblastoma, similar a lo reportado por Pang et al.3, y no cumpliéndose en nuestra serie la posible relación entre la positividad y los casos más graves como señala recientemente Pranzatelli34. Al contrario que en otros artículos, no encontramos presencia de anticuerpos antineuronales23,34–37.
Curso evolutivo, recaídas y secuelasEl tiempo medio necesario para alcanzar una completa resolución de los síntomas (11,72 meses) fue similar a otros estudios como el de Huddar et al.23 (12 meses). La duración media de mantenimiento del tratamiento en nuestros pacientes fue de 17,6 meses, algo inferior a lo referido en otras series como la del propio Huddar et al.23 (22,3 meses ± 20 meses; tres meses a cinco años), aunque con una tasa de recaídas de 43% (6/14), superior a la nuestra (20%). En este sentido pueden haber influido otros factores descritos en líneas anteriores, como el mayor retraso en el diagnóstico y en la instauración del tratamiento o, en relación con nuestra muestra un tiempo de seguimiento insuficiente en algunos casos y las pautas de inmunoterapia más incisivas y precoces que empleamos desde los últimos años.
Aunque no hay un tiempo estipulado, suele considerarse recaída si los síntomas de SOMA reaparecen y se mantienen durante al menos 48-72 horas; mientras que se habla de pseudo-recaída ante empeoramientos transitorios de los síntomas después de ciertas situaciones como una sedación o anestesia sin que exista progresión38. De este modo, el curso del SOMA puede ser monofásico o multifásico31 y sin un periodo de seguimiento prolongado no es posible diferenciar con seguridad que pacientes mantendrán su remisión y cuales recaerán31.
En nuestra serie, 11 de los 20 pacientes presentaron un curso monofásico, sin reaparición durante el seguimiento (de entre dos y 7,5 años). Cuatro casos (20%) presentaron un curso claramente remitente-recurrente, tres de ellos con una sola recaída y uno con dos. Tres se asociaron al descenso de la corticoterapia, que es el desencadenante más habitual, seguido de las infecciones39,40. Un paciente recayó a los dos años de suspender la inmunoterapia. En este caso, el amplio lapso asintomático y sin tratamiento, apoya la necesidad de un seguimiento prolongado para definir el curso de la enfermedad, la heterogeneidad de las recaídas y el tiempo de evolución en que ocurren31. En este sentido se han documentado recaídas incluso en la edad adulta41. Desde el punto de vista del régimen de inmunoterapia previo, las recaídas aparecieron en mayor proporción en pacientes previamente tratados con monoterapia (2/6) que en los que habían recibido politerapia (2/14), lo que apoya el uso del tratamiento combinado, tal y como preconizan las tendencias actuales38. Destacamos que dos de los cuatro pacientes que presentaron recaídas sufrieron secuelas cognitivas, siendo su minimización uno de los objetivos de los nuevos regímenes terapéuticos.
Dos casos basalmente asintomáticos, presentan síntomas mínimos puntualmente (movimientos oculares inespecíficos o ligera ataxia coincidiendo con infecciones) que se autolimitan sin realizar cambios terapéuticos. Probablemente estas exacerbaciones encajan más con el concepto de pseudo-recaída. Su presencia puede reflejar la no consecución de un control completo de la enfermedad, con un aumento del riesgo de una verdadera recaída38,40, haciendo necesario el seguimiento prolongado del paciente.
La evolución a largo plazo de los pacientes con SOMA está marcada principalmente por las secuelas neuropsicológicas, los trastornos de conducta y las dificultades de aprendizaje; mientras que los síntomas neurológicos iniciales tienden a mejorar con la inmunoterapia33 o incluso espontáneamente. En nuestra serie lo más frecuente fue el desarrollo evolutivo de TDAH y/o trastorno del aprendizaje de la lectoescritura, no observándose un patrón de deterioro progresivo con la edad a diferencia de lo referido por Mitchell42, quien planteaba la posibilidad de una «encefalopatía progresiva» en lugar de un «insulto» limitado en el tiempo.
Estudio de posibles condicionantes pronósticosAunque existen casos infrecuentes de resolución espontánea3,22, es preciso un tratamiento inmunomodulador. En los últimos años se ha producido un cambio en los regímenes de tratamiento, que abogan por la intensificación precoz del mismo, instaurando una biterapia desde el principio (generalmente esteroides e inmunoglobulinas) y asociando un fármaco de segunda línea precozmente si la respuesta no es satisfactoria43. Incluso algunos autores plantean la triterapia inicial44. El objetivo de la optimización del tratamiento (y con ello mejor control de la base inmunopatológica de la enfermedad) no es sólo la resolución de la clínica neurológica aguda, sino minimizar las repercusiones cognitivas permanentes.
Aunque una proporción de pacientes mantienen un desarrollo normal tras haber sufrido un SOMA, la mayoría muestran ciertos déficit cognitivos o neuropsicológicos (afectándose atención, memoria, visomotor, memoria de trabajo) a largo plazo, generalmente con un pronóstico motor normal o casi normal24. Series publicadas previamente reportan algún déficit neuropsicológico en el 80% de los pacientes investigados45, aunque a la hora de interpretar esta información se debe tener en cuenta la naturaleza retrospectiva de la mayoría de las series y la diversidad de tratamientos empleados. Factores como la menor edad al diagnóstico, el retraso en el tratamiento inmunosupresor, la presentación clínica inicial más grave, o la reaparición de la clínica tras el descenso de la corticoterapia (y el consecuente retraso en la resolución clínica completa) se han discutido como posibles factores de mal pronóstico21,24.
En nuestra muestra, no hubo diferencias significativas en la frecuencia de afectación cognitiva a los dos años del debut entre aquellos de inicio clínico precoz (anterior a los 18 meses) y los de inicio posterior.
Por otro lado, aunque no hemos encontrado una relación significativa entre precocidad de inicio de la inmunoterapia y evolución de la gravedad clínica, sí que se asoció con una menor frecuencia de afectación cognitiva a los dos años, que consideramos clínicamente relevante. De igual forma, aunque no se hallaron diferencias significativas en cuanto a afectación cognitiva entre el grupo que precisó fármacos de segunda línea frente a los que no, todos los pacientes que iniciaron triterapia en los primeros seis meses de tratamiento mantuvieron un estado cognitivo normal a los dos años del debut, a diferencia de los que la iniciaron después. Estos datos nos llevan a señalar la importancia de la precocidad del tratamiento, así como la consideración de la triterapia desde los primeros meses.
En nuestra serie la media de tiempo hasta la instauración de la inmunoterapia es de aproximadamente 1,1 meses desde el debut clínico y se aproxima a 0,3 meses desde el diagnóstico definitivo en los casos de debut más reciente. En todo caso podríamos decir que, en nuestro medio y en la actualidad parece cumplirse la tendencia a la implementación precoz de la inmunoterapia, con tiempos (aproximados) inferiores a lo descrito en otras series como la de Huddar et al.23 (tiempo medio desde diagnóstico hasta inicio de inmunoterapia de 6,1 ± 7,5 meses; rango dos a 24 meses).
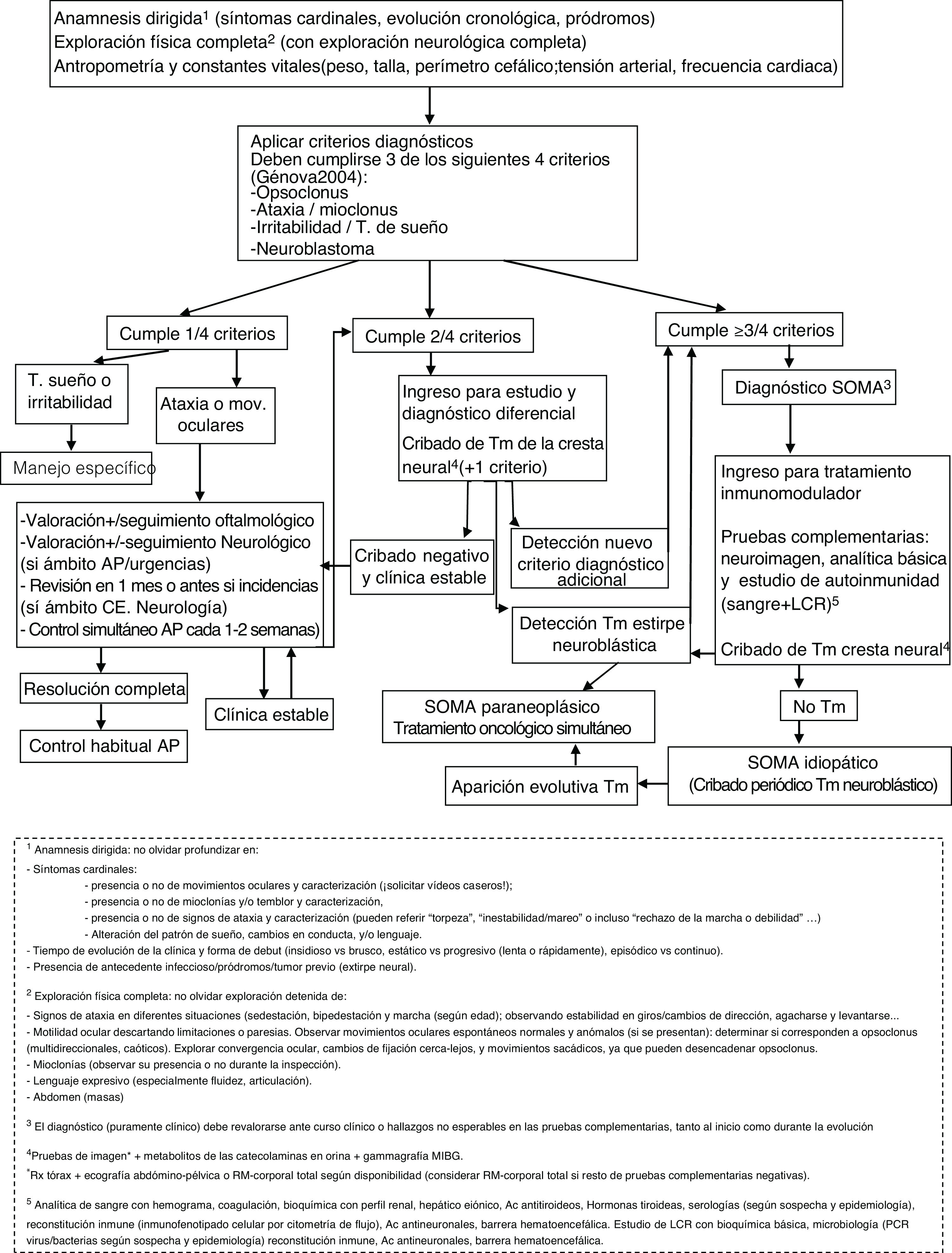
ConclusiónAunque con la limitación del pequeño tamaño de la muestra, la naturaleza retrospectiva y la ausencia de un protocolo de manejo común, en nuestra serie parece existir cierta relación entre los regímenes terapéuticos y la evolución neurocognitiva. En este sentido, el primer requisito es un diagnóstico temprano, siendo imprescindible el reconocimiento de la clínica. Destacamos la importancia de la búsqueda activa de otros síntomas y signos sugestivos (a veces sólo conductuales) entre aquellos pacientes que debutan con ataxia, ante los cuales se debe mantener un alto nivel de sospecha. De nuevo, se documenta una importante asociación a tumores neuroblásticos, generalmente poco agresivos, en cuya búsqueda parece imprescindible el uso de técnicas de imagen como la TC o la RM, dado el alto porcentaje de falsos negativos en otras pruebas que dependen de la actividad metabólica o el tamaño tumoral (figura 1, algoritmo diagnóstico ante posible SOMA). La aparición o no de secuelas podría haberse visto influida por la intensificación adecuada de la inmunoterapia cuando la respuesta no fue completa, así como por su precocidad. Por tanto, subrayamos la importancia del tratamiento combinado desde el inicio, así como la triterapia ante una respuesta insuficiente (figura 2, algoritmo terapéutico en SOMA). En este sentido, creemos que el desarrollo e incorporación a la neuroinmunología infantil de tratamientos más dirigidos frente al mecanismo inflamatorio teórico subyacente como el rituximab, ha supuesto un cambio muy positivo. El estudio e incorporación de nuevos marcadores inmunológicos y de tratamientos dirigidos, así como su protocolización, podría ayudar en la búsqueda de un manejo y pronóstico óptimo en el SOMA.
; AP (Atención Primaria); CE (Consultas Externas); LCR (líquido cefalorraquídeo; mov (movimientos); MIBG (metayodobencilguanidina); PCR (proteína C reactiva); RM (resonancia magnética); Rx (radiografía); SOMA (Síndrome opsoclonus mioclonus).")
Algoritmo diagnóstico ante posible SOMA. Ac (anticuerpos); AP (Atención Primaria); CE (Consultas Externas); LCR (líquido cefalorraquídeo; mov (movimientos); MIBG (metayodobencilguanidina); PCR (proteína C reactiva); RM (resonancia magnética); Rx (radiografía); SOMA (Síndrome opsoclonus mioclonus).
; DXM (dexametasona); FC (frecuencia cardiaca); g (gramos); Ig (inmunoglobulina); iv (intravenosa); Kg (kilogramo), m2 (metro cuadrado); mg (miligramos); p.ej. (por ejemplo); RI (reconstitución inmune); RTX (rituximab); TA (tensión arterial).")
Algoritmo terapéutico en SOMA. CFM (ciclofosfamida); DXM (dexametasona); FC (frecuencia cardiaca); g (gramos); Ig (inmunoglobulina); iv (intravenosa); Kg (kilogramo), m2 (metro cuadrado); mg (miligramos); p.ej. (por ejemplo); RI (reconstitución inmune); RTX (rituximab); TA (tensión arterial).
La presente investigación no ha recibido ninguna beca específica de agencias de los sectores público, comercial, o sin ánimo de lucro.
Conflictos de interésLos autores declaran no tener ningún conflicto de interés.

