




Paraneoplastic neurological syndromes (PNS) are a heterogeneous group of conditions attributable to cancer that do not result from local or metastatic presence of tumour cells. They may affect any area of the central nervous system, and are classified as classical (encephalitis, encephalomyelitis, subacute cerebellar degeneration, sensory neuropathy, myasthenic syndrome, dermatomyositis, and opsoclonus-myoclonus syndrome) and non-classical. PNS continue to be a diagnostic challenge; diagnosis is based on the criteria established by Graus et al.1 in 2004 (Table 1). Regarding aetiology, most neurological symptoms result from CD8+ T cell cytotoxicity.
Diagnostic criteria for paraneoplastic neurological syndromes (PNS) established by Graus et al.1.
| Definite PNS |
| 1. A classical syndrome and cancer, with or without onconeural antibodies, that develops within 5 years of the diagnosis of the neurological disorder. |
| 2. A non-classical syndrome that resolves or significantly improves after cancer treatment without concomitant immunotherapy, provided that the syndrome is not susceptible to spontaneous remission. |
| 3. A non-classical syndrome with onconeural antibodies (well-characterised or not) and cancer that develops within 5 years of the diagnosis of the neurological disorder. |
| 4. A neurological syndrome (classical or not) with well-characterised onconeural antibodies (anti-Hu, -Yo, -CV2, -Ri, -Ma2, or -amphiphysin), and no cancer. |
| Possible PNS |
| 1. A classical syndrome, no onconeural antibodies, no cancer but at high risk to have an underlying tumour. |
| 2. A neurological syndrome (classical or not) with partially-characterised onconeural antibodies and no cancer. |
| 3. A non-classical syndrome, no onconeural antibodies, and cancer present within 2 years of diagnosis. |
We present an extremely unusual case of PNS in a child aged 2 years and 8 months. She was attended at our department due to somnolence and gait instability of 20 hours’ progression. She presented no other symptoms and had no history of head trauma or toxic substance use. The physical examination detected somnolence with response to stimulation (Glasgow Coma Scale score of 14/15), irritability, ataxic gait, and gaze-induced nystagmus. The rest of the examination identified no further alterations.
We started empirical antibiotic therapy; biochemical analysis, a complete blood count, acute-phase reactant quantification, and a blood culture yielded normal results.
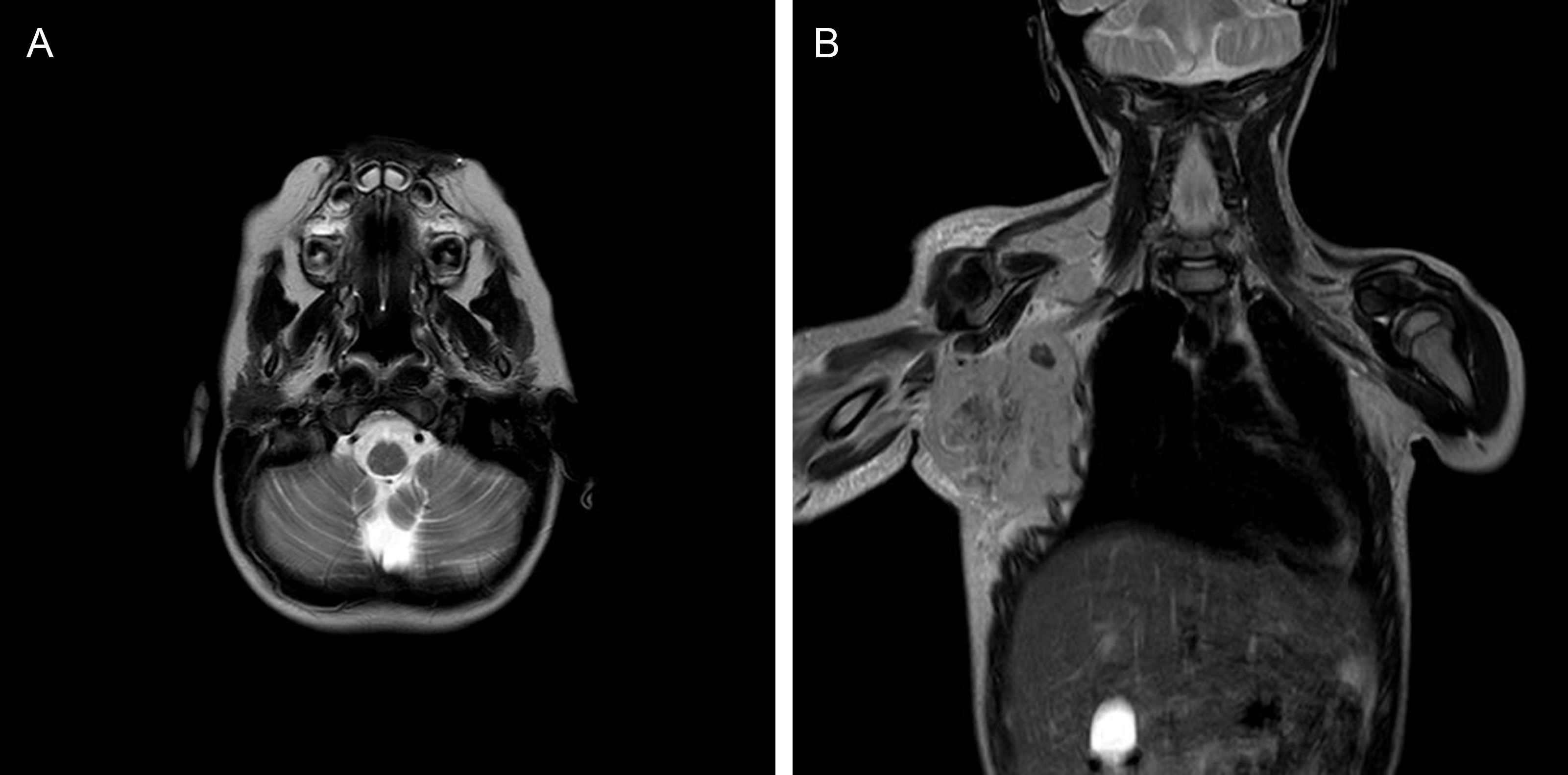
A CT scan and a lumbar puncture with CSF cytobiochemical analysis, multiplex polymerase chain reaction assay, and cultures detected no abnormal findings. A brain MRI scan (Fig. 1A) revealed signs compatible with acute cerebellitis. Immunomodulatory treatment with intravenous immunoglobulins and megadose corticosteroids achieved a progressive improvement with complete symptom resolution. Tests for antineuronal antibodies (anti-NMDAR and anti-MOG antibodies) in the blood and CSF yielded negative results.
 Brain MRI showing lesions compatible with cerebellitis. (B) Chest MRI showing a large bulky mass on the right side.")
Three months after diagnosis, with the patient presenting no neurological symptoms, we detected enlarged lymph nodes in the right axillary, epitrochlear, and infraclavicular regions; the adenopathies were hard and adhered to deep tissue, in association with a large bulky mass (Fig. 1B). A lymph node biopsy revealed alveolar rhabdomyosarcoma with neuroendocrine differentiation (CD56+) and positive PAX3-FOXO1 translocation.
A more extensive study detected antineuronal cell-surface antibodies (mGluR2) in the blood and CSF; these had not previously been detected. This finding was confirmed by positive immunoreactivity in a culture study of live hippocampal neurons. Tumour cells also expressed mGluR2. These findings supported a diagnosis of paraneoplastic cerebellitis. Chemotherapy was started following the EpSSG RMS2005 protocol for very high-risk patients, with good response, enabling tumour resection.
PNS are rare in the general population, presenting in fewer than 1% of patients with cancer; the incidence rate in children is unknown.2 The tumours most frequently involved in PNS are neuroendocrine tumours (small-cell lung cancer, neuroblastoma), tumours affecting organs with immunoregulatory functions (thymoma), or those containing mature or immature neural tissue (teratoma)2. In paediatric patients, the most frequent PNS is opsoclonus-myoclonus syndrome, which is associated with neuroblastoma in 50% of cases.3
Rhabdomyosarcoma accounts for 5%-8% of all tumours in paediatric patients. It has been associated with hypercalcaemia, bone alterations,3 and vasculitis in paediatric patients.4 To date, only one case has been reported of PNS associated with rhabdomyosarcoma with neuroendocrine differentiation and positive anti-Hu antibodies, in an adult patient5; no cases have been reported in children.
The characterisation of onconeural antibodies is essential in these syndromes, as they are associated with a specific type of tumour. At present, the most frequently studied antibodies are anti-Hu, -Yo, -Ma2, -CRMP-5, -amphiphysin, and -Ri antibodies.6 Presence of these well-characterised antibodies together with classical neurological symptoms is sufficient to determine the specific paraneoplastic syndrome.1
We describe a new antibody targeting surface mGluR2 in a patient with ataxia and cancer. Although classic neoplastic cerebellitis is associated with onconeural antibodies against intracellular antigens, our patient’s symptoms improved with immunotherapy, which suggests that anti-mGluR2 antibodies played a pathogenic role.
Most PNS resolve with treatment of the underlying neoplasia, with some exceptions, as with anti-NMDAR encephalitis, where treatment of the tumour does not alter the course of the neurological disease. Immunosuppressive therapy achieves varying rates of response; good response to this treatment has been observed in patients with anti-GABABR and anti-AMPAR antibodies.3
Please cite this article as: Fernández Escobar V, del Pozo Carlavilla M, Ruiz Garcia R, Buedo Rubio MI. Síndrome paraneoplásico cerebeloso secundario a rabdomiosarcoma: una etiología muy poco frecuente. Neurología. 2021;36:645–646.
